
Simultaneous Detection of Actinobacillus pleuropneumoniae and Haemophilus parasuis in Pig by Duplex Droplet Digital PCR
- DOI
- 10.2991/efood.k.200918.001How to use a DOI?
- Keywords
- Droplet digital PCR; detection; pig; Actinobacillus pleuropneumoniae; Haemophilus parasuis
- Abstract
Actinobacillus pleuropneumoniae (A. pleuropneumoniae) and Haemophilus parasuis (H. parasuis) are important pathogens of swine, which cause great economic losses in the swine industry and bring great safety hazards to food safety. In this study, we established a duplex Droplet Digital Polymerase Chain Reaction (ddPCR) assay for sensitive detection of A. pleuropneumoniae and H. parasuis simultaneously in pork. The limit of detection of plasmid DNA could reach 2 copies/μL for both A. pleuropneumoniae and H. parasuis; which were approximately 50- and 20-fold greater sensitivity than Quantitative real-time PCR (qPCR). Both qPCR and ddPCR showed high linearity and positive correlation with standards. The detection results of clinical suspected samples for A. pleuropneumoniae and H. parasuis indicated the positive detection rate of ddPCR (10.7% and 12.5%) was higher than that of qPCR (7.1% and 8.9%). Therefore, the duplex ddPCR assay could be used as an effective quantitative detection to diagnose A. pleuropneumoniae and H. parasuis simultaneously, which may lay a foundation for the development of substitute rapid diagnostic technique.
- Graphical Abstract
- Copyright
- © 2020 The Authors. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
As an important source of protein intake and a national strategic reserve, pork has high nutritional value and is one most important red meat foods in the human diet, and the demand for pork is increasing globally [1]. In recent years, people pay more attention to food safety, and the safety of meat has always been the focus of societal concerns. The serious meat safety issues may directly lead to the health problems of consumers, and the government needs to recall the contaminated products on the market. When the swine occasionally suffer from certain major diseases, it often changes the demand for pork and further affects the price of pork [2]. Such issues like the outbreak of Streptococcus suis in Sichuan province [3], the swine flu swept the whole world [4], and the African swine fever broke out in China [5] happened these years, which caused a serious blow to the swine industry. Then the quality and safety of pork closely related to human health cannot control effectively. It is therefore critical for timely monitoring and prevention of swine epidemics to ensure the safety of pork consumption.
Actinobacillus pleuropneumoniae (A. pleuropheumoniae) is a respiratory pathogen in swine, and it is the pathogenic agent of porcine pleuropneumonia in swine [6]. Haemophilus parasuis (H. parasuis) is the specific pathogenic cause of Glässer’s disease in swine, which brings about polyserositis in swine, including pleuritis, peritonitistis and arthritis [7]. Both of them are among the most common bacterial causes of porcine respiratory disease, and often associated with the porcine respiratory disease complex caused by both primary and secondary respiratory pathogens [8]. Although these two pathogens will not affect the food safety directly, the present of A. pleuropheumoniae and H. parasuis in pig can damage the quality and taste of meat, and reduce the edible quality of pork. Furthermore, the porcine respiratory disease is associated with a high incidence rate and fatality rate, which causes substantial losses in the swine industry worldwide and the associated healthcare costs [9–11].
In recent years, Enzyme-linked Immunosorbent Assay (ELISA) [12,13], nested Polymerase Chain Reaction (nested PCR) [14,15] and Quantitative real-time PCR (qPCR) [16–18] have been developed to detect and serotype H. parasuis and A. pleuropneumoniae with improved sensitivity and efficiency. However, it is difficult to distinguish the mixed infection of these two diseases by clinical symptoms and pathological features in clinical diagnosis. Thus, multiplex PCR detection has been widely used in many fields in recent years, such as monitoring food pathogenic microorganisms [19,20] and detecting veterinary diagnosis [21–23]. Moreover, Droplet Digital PCR (ddPCR), the latest version of digital PCR, allows better accuracy of DNA quantification [24]. The ddPCR can detect bacterial pathogens with greater sensitivity, which is crucial for early diagnosis and disease control. At present, with the use of commercial ddPCR system such as the Bio-Rad QX100/200, many studies involving duplex quantitative detection have been carried out [25–27]. In this study, we established a duplex ddPCR assay for identifying A. pleuropneumoniae and H. parasuis simultaneously in comparison with qPCR.
2. MATERIALS AND METHODS
2.1. Strains and Clinical Samples
Field isolates of A. pleuropneumoniae strains (serotype 1–15), field isolates of H. parasuis strains (serotype 1–15), Classical Swine Fever Virus (CSFV), Porcine Circovirus 2 (PCV2), Pseudorabies Virus (PRV), Porcine Reproductive and Respiratory Syndrome Virus (PRRSV), Porcine Epidemic Diarrhea Virus (PEDV), Streptococcus suis (S. suis), Staphylococcus aureus (S. aureus) (ATCC25923), Salmonella Typhimurium (ATCC14028) were obtained from a commercial company (Yongshun Biological Pharmaceutical, Guangdong, China). Fifty lung tissues from 50 healthy piglets in one pig farm and 140 clinical tissue samples (lungs and tonsils) from 10 pig farms were collected from the Institute of Animal Sciences (Guangdong Academy of Agricultural Sciences, Guangdong, China). All clinical samples were stored at −80°C for further use.
2.2. Nucleic Acid Extraction and Reverse Transcription
The DNA of A. pleuropneumoniae, H. parasuis, PCV2, PRV, S. suis, S. aureus, Salmonella and the RNA of CSFV, PRRSV and PEDV was extracted by DNA/RNA extraction kit (Magen Bio-tech, Shanghai, China), according to the manufacturer’s instruction. And the RNA was reverse transcribed using the PrimerScript RT reagent kit (TaKaRa, Dalian, China). All DNA and cDNA were stored at −20°C.
2.3. Primers and Probes
The primers and probes of qPCR and ddPCR assays were designed by using Oligo Primer Analysis Software (Molecular Biology Insights, Colorado Springs, CO, USA) then synthesized from a commercial company (Sangon Biotech, Shanghai, China). The probes were labeled with 6-carboxy-fluorescein (FAM) and VIC as the fluorescent reporter, respectively; and minor groove binder (MGB) as the fluorescence quencher. The primers and probe set for A. pleuropneumoniae were designed were as follow: F1 (5′-GCTGACCGCAGTATAACTGTATGG-3′); R1 (5′-CGTCCCCAGTCGTTGATATTAT-3′); and probe (5′-FAM-ATTATTTGGCACTGACGGTG-MGB-3′); for H. parasuis were F1 (5′-TCGCCTTGGCCGTAATTCTA-3′); R1 (5′-ACGTAAGCCTTTTCTGTTGTAACATC-3′); and probe (5′-VIC-AAATGATGCAGGATGGG-MGB-3′). The plasmid standards pUC57-APP of A. pleuropneumoniae and pUC57-HPS of H. parasuis were constructed by inserting the 551 bp sequence of apxIVA (GenBank No. CP030753.1) amplified by primers: FP: 5′-TTGCTGACCGCAGTATAACTGTATG-3′; RP: 5′-GAGGTAAAACAATATATCAATAGCTTAAC-3′, and 747 bp sequence of OMP P2 (GenBank No. KU508604.1) amplified by primers: FP: 5′-CTACGGTTTTGGTCGTTATGAG-3′; RP: 5′-GCAGACTATAAATTACATAAACAAGTTGT-3′ into commercial vector pUC57 (Sangon Biotech, Shanghai), respectively. And the plasmid pUC57-APP and pUC57-HPS were used as the positive, respectively. The purified recombinant plasmids were quantified (Quibt 3.0; Thermo Fisher Scientific, Waltham, MA, USA), and recalculated to plasmid copies/μL using an online calculator (available at http://scienceprimer.com/copy-number-calculator-for-realtime-pcr). The recombinant plasmids were serially 10-fold and twofold diluted. Then, the dilutions and plasmids were stored at −20°C and −70°C, respectively.
2.4. ddPCR Assay
A duplex ddPCR assay was designed to amplify A. pleuropneumoniae and H. parasuis with primers and probes by using QX200 droplet digital PCR system (Bio-Rad Laboratories, Hercules, CA, USA). In each step of amplification, the optimized 20 μL mixtures of duplex ddPCR reaction containing 10 μL of 2 × ddPCR supermix for probes (no dUTP; Bio-Rad), 800 nM of each primer, 400 nM of each TaqMan-MGB probes, and 1 μL of each DNA template. 20 μL of ddPCR reaction mixtures and 70 μL of Droplet Generation oil for Probes (Bio-Rad) were inserted in an eight-well cartridge to generate the droplets. With the use of a droplet generator (Bio-Rad), each sample was divided into 20,000 water-in-oil nanolitre-sized droplets. After that, the 40 μL generated droplet emulsion was transferred to a new 96-well PCR plate (Eppendorf, Hauppauge, NY, USA), then heat-sealed with a pierce-able sealing foil sheet by using the PX1™ PCR plate sealer (Bio-Rad) and amplified in C1000 Touch™ deep-well thermal cycler (Bio-Rad) finally. The optimized cycling conditions of ddPCR were as follows: 95°C for 10 min; then 40 cycles of 94°C for 30 s, 56.3°C for 60 s, and one cycle of 98°C for 10 min, and ending at 12°C.
2.5. qPCR Assay
The qPCR assay was performed using ABI QuantStudio 6 Flex real-time PCR system (Thermo Fisher Scientific). The 20 μL qPCR reactions comprised 10 μL of AceQ qPCR probe master mix (Vazyme, Nanjing, China), 1 μL of each DNA template, and the final concentrations of primer and probe were 200 and 100 nM, respectively. The reaction conditions of qPCR were as follows: 95°C for 5 min, followed by 35 cycles at 95°C for 10 s, and 60°C for 34 s.
2.6. Specificity and Reproducibility Test of ddPCR
The specificity of the ddPCR assay was validated, and nucleic acid extracts of A. pleuropneumoniae, H. parasuis, PCV2, PRV, S. suis, S. aureus, Salmonella, CSFV, PRRSV and PEDV were tested.
The robustness and reproducibility of the qPCR and ddPCR were determined by serially diluted recombinant plasmids. To evaluate intra- and inter-assay reproducibility, each template was tested in triplicate, and the Standard Deviation (SD) and the Coefficient of Variation (CV) were calculated and analyzed statistically.
2.7. Comparison of ddPCR and qPCR Assay
The pUC57-APP and pUC57-HPS which serially 10-fold and twofold diluted were used to compare the sensitivity and accuracy between ddPCR and qPCR. The quantitative agreement between ddPCR and qPCR measurements was access by correlation analysis using standard curves of qPCR and ddPCR.
2.8. Clinical Sample Detection
One hundred and forty clinical samples and 50 lung tissue samples were used to evaluate the ability of qPCR and ddPCR. The positive detection rate was compared and evaluated the sensitivity of both methods.
2.9. Statistical Analysis
The correlations and regressions analysis of the standard curves from qPCR were analyzed by origin 2017 (OriginLab, MA, USA). The copy number of the initial templates from ddPCR was analyzed by QuantaSoft analysis software (Bio-Rad). Kappa statistics were used to evaluate the agreement of clinical detection results between qPCR and ddPCR.
3. RESULTS
3.1. Primer Annealing Temperature Optimizing
According to the manufacturer’s instructions, the optimized annealing temperature of qPCR was 60°C. For ddPCR assay, the annealing temperature gradients test was carried out at the following temperatures: 60°C, 59.4°C, 58.3°C, 56.3°C, 53.9°C, 52°C, 50.7°C, and 50°C. As shown in Figure 1, the optimal annealing temperature of ddPCR was 56.3°C, which could effectively distinguish the signals between fluorescent channels peaked.
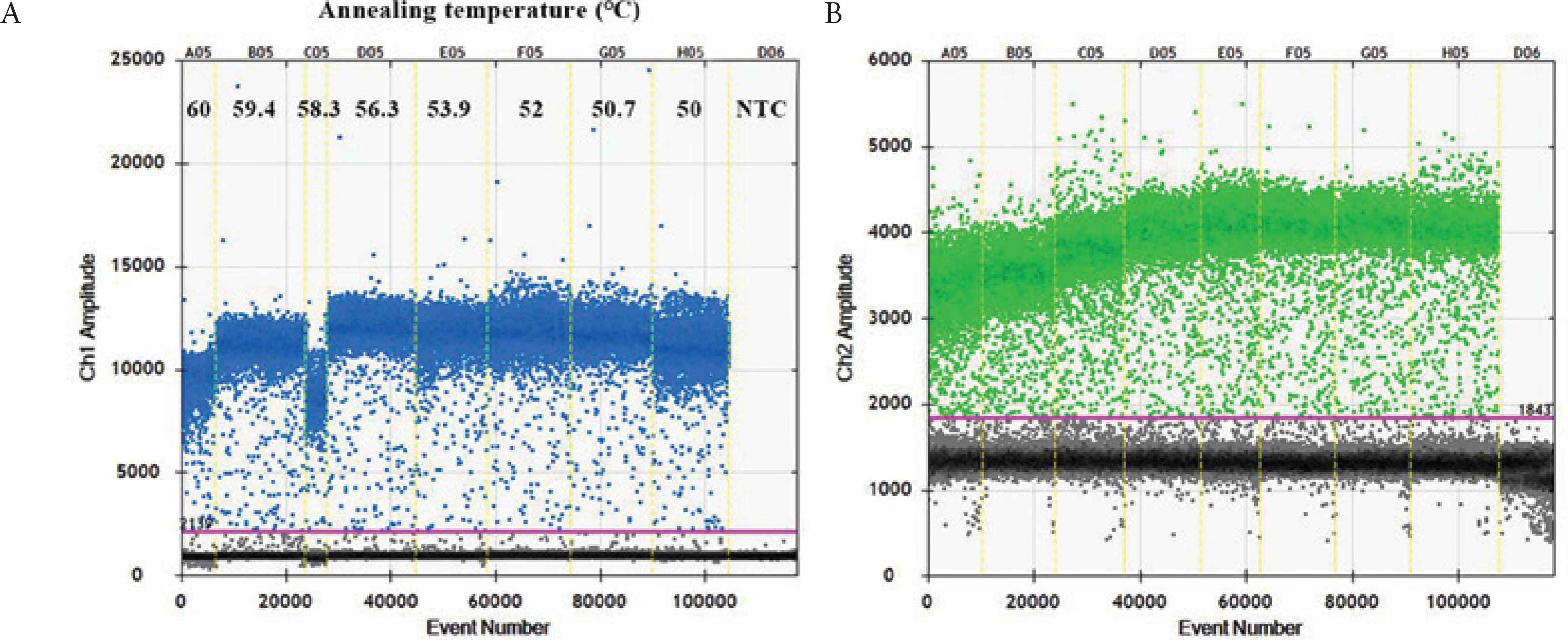
Optimization of annealing temperature for detection of pUC57-APP and pUC57-HPS. (A) Lane A05:60°C; lane B05:59.4°C; lane C05:58.3°C; lane D05:56.3°C; lane E05:53.9°C; lane F05:52°C; lane G05:50.7°C; lane H05:50°C; lane D06: NTC. Blue plots indicate pUC57-APP (FAM signal). (B) Green plots indicate pUC57-HPS (VIC signal).
3.2. Specificity and Reproducibility Test of ddPCR
The CV is used to reflect the robustness and reproducibility of ddPCR. As shown in Tables 1 and 2, CV values of intra-assay variation was 0.54–6.67% for A. pleuropneumoniae and 0.64–6.83% for H. parasuis, and inter-assay variation was 0.28–5.33% for A. pleuropneumoniae and 0.85–4.76% for H. parasuis, which indicated good reproducibility and robustness of ddPCR. The results of specificity test showed the target strains and positive samples were correctly identified and no generating false-positive or false-negative results were detected. Thus, it confirmed that the primers and probes designed in this study had no non-specific amplification in the genomes on the above pathogens, which indicated that ddPCR had good specificity.
| Concentration of pUC57-APP (copies/μL) | Intra-assay variation | Inter-assay variation | ||||
|---|---|---|---|---|---|---|
| Mean (copies/μL) | SD | CV (%) | Mean (copies/μL) | SD | CV (%) | |
| 1.4 × 105 | 7441 | 40.45 | 0.54 | 7457 | 20.79 | 0.28 |
| 1.2 × 104 | 875 | 6.66 | 0.76 | 882 | 12.34 | 1.40 |
| 1.1 × 103 | 102 | 6.56 | 6.43 | 101 | 4.51 | 4.48 |
| 95 | 14 | 0.75 | 5.20 | 16 | 0.55 | 3.52 |
| 46 | 7 | 0.35 | 4.92 | 7 | 0.31 | 4.09 |
| 22 | 2 | 0.15 | 6.67 | 2 | 0.12 | 5.33 |
Robustness and reproducibility of ddPCR assay for A. pleuropneumoniae
| Concentration of pUC57-HPS (copies/μL) | Intra-assay variation | Inter-assay variation | ||||
|---|---|---|---|---|---|---|
| Mean (copies/μL) | SD | CV (%) | Mean (copies/μL) | SD | CV (%) | |
| 4.8 × 104 | 4863 | 31.00 | 0.64 | 4874 | 43.84 | 0.90 |
| 3.4 × 103 | 746 | 26.69 | 3.58 | 776 | 6.56 | 0.85 |
| 2.4 × 102 | 115 | 5.69 | 4.93 | 137 | 3.06 | 2.22 |
| 17 | 17 | 0.96 | 5.57 | 18 | 0.59 | 3.18 |
| 8 | 8 | 0.51 | 6.47 | 8 | 0.35 | 4.30 |
| 3 | 2 | 0.16 | 6.83 | 2 | 0.10 | 4.76 |
Robustness and reproducibility of ddPCR assay for H. parasuis
3.3. Analysis of Standard Curves and Detection Limits
To assess the accuracy and sensitivity, serial dilutions of recombinant pUC57-APP and pUC57-HPS were tested by both PCR assays (Table 3 and Figure 2). Standard curves and efficacies of qPCR were obtained upon serial 10-fold dilutions. A. pleuropneumoniae and H. parasuis PCR efficacies were 1.064 and 1.143, respectively. The Limit of Detection (LoD) of duplex qPCR was 105 and 45 copies/μL for plasmid DNA of A. pleuropneumoniae and H. parasuis, respectively. In contrast, the LoD for ddPCR detection of plasmid was 2 copies/μL for both A. pleuropneumoniae and H. parasuis. The LoD for ddPCR detection of A. pleuropneumoniae and H. parasuis was 50-fold and 20-fold better than qPCR, respectively. The correlation coefficient (R2) was calculated on the mean value of target copy numbers measured in the sensitivity of ddPCR and qPCR, which is used to determine the linearity over the dynamic range. The results were showed that, in the range of quantification, both qPCR and ddPCR have good linearity (Figure 3).
| Concentration of plasmidsa (copies/μL) | qPCR (Mean Cq value) | ddPCRb (Mean concentration, copies/μL) | |||
|---|---|---|---|---|---|
| pUC57-APP | pUC57-HPS | pUC57-APP | pUC57-HPS | pUC57-APP | pUC57-HPS |
| 1.6 × 106 | 6.8 × 105 | 21.4 | 20.5 | NT | NT |
| 1.4 × 105 | 4.8 × 104 | 24.6 | 23.7 | 7441 | 4863 |
| 1.2 × 104 | 3.4 × 103 | 28.0 | 27.4 | 875 | 746 |
| 1.1 × 103 | 2.4 × 102 | 31.3 | 31.1 | 102 | 115 |
| 95 | 17 | 34.0 | 35.1 | 14 | 17 |
| 46 | 8 | ND | ND | 7 | 8 |
| 22 | 3 | ND | ND | 2 | 2 |
| NTC | NTC | ND | ND | ND | ND |
NTC, no template control; ND, not detected; NT, not tested; Cq, quantification cycle.
Concentration based on 10-fold and twofold serial dilutions of plasmid.
Concentration based on ddPCR detection.
Comparative limit of detection between ddPCR and qPCR assay
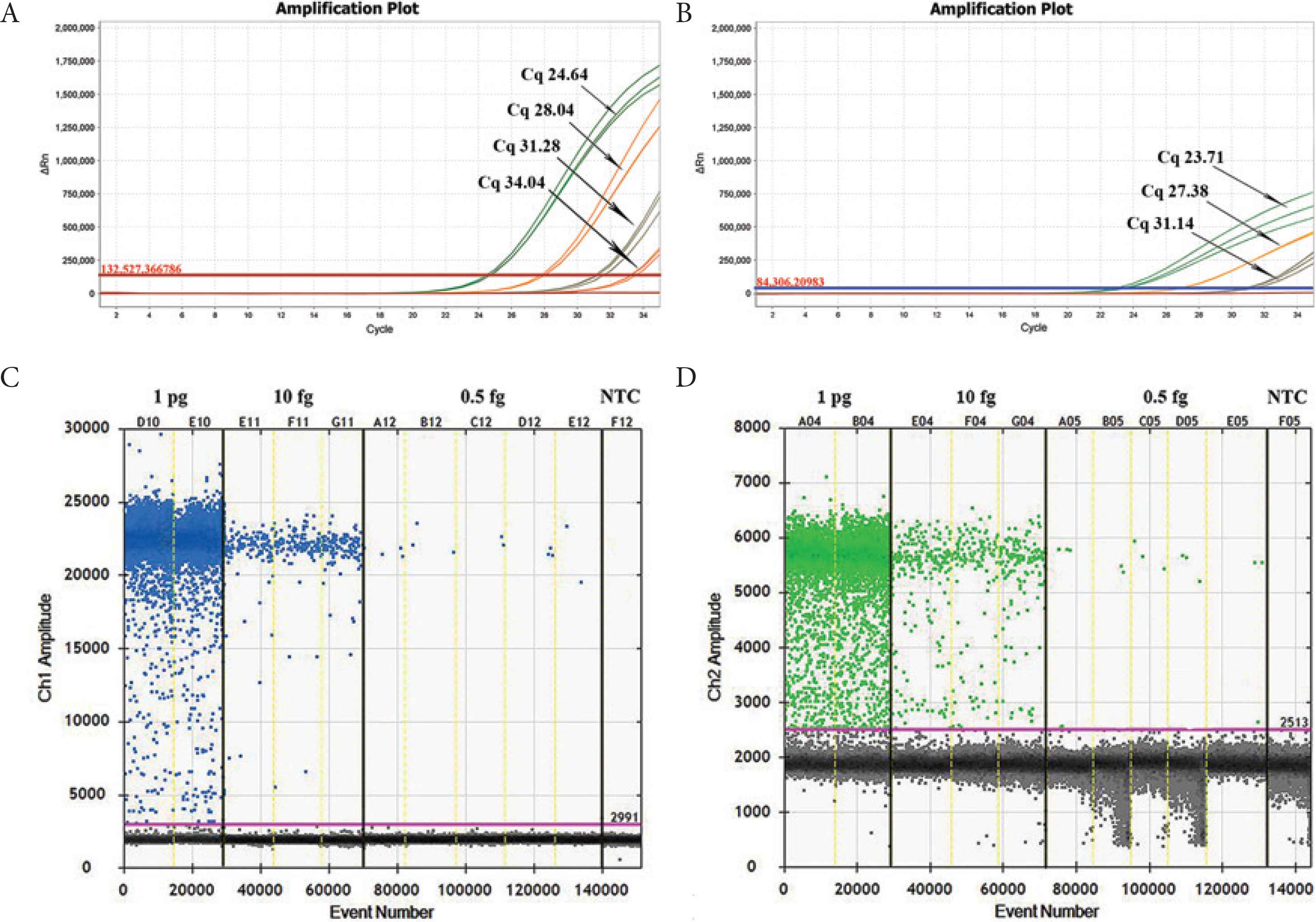
Comparison of the sensitivity of pUC57-APP and pUC57-HPS by RT-PCR and ddPCR. (A) and (B) The real-time fluorescence monitoring of pUC57-APP and pUC57-HPS were performed by qPCR, respectively. (C) and (D) The fluorescence amplitude of pUC57-APP and pUC57-HPS by ddPCR (1D Droplet Plots), respectively. NTC, negative template control.
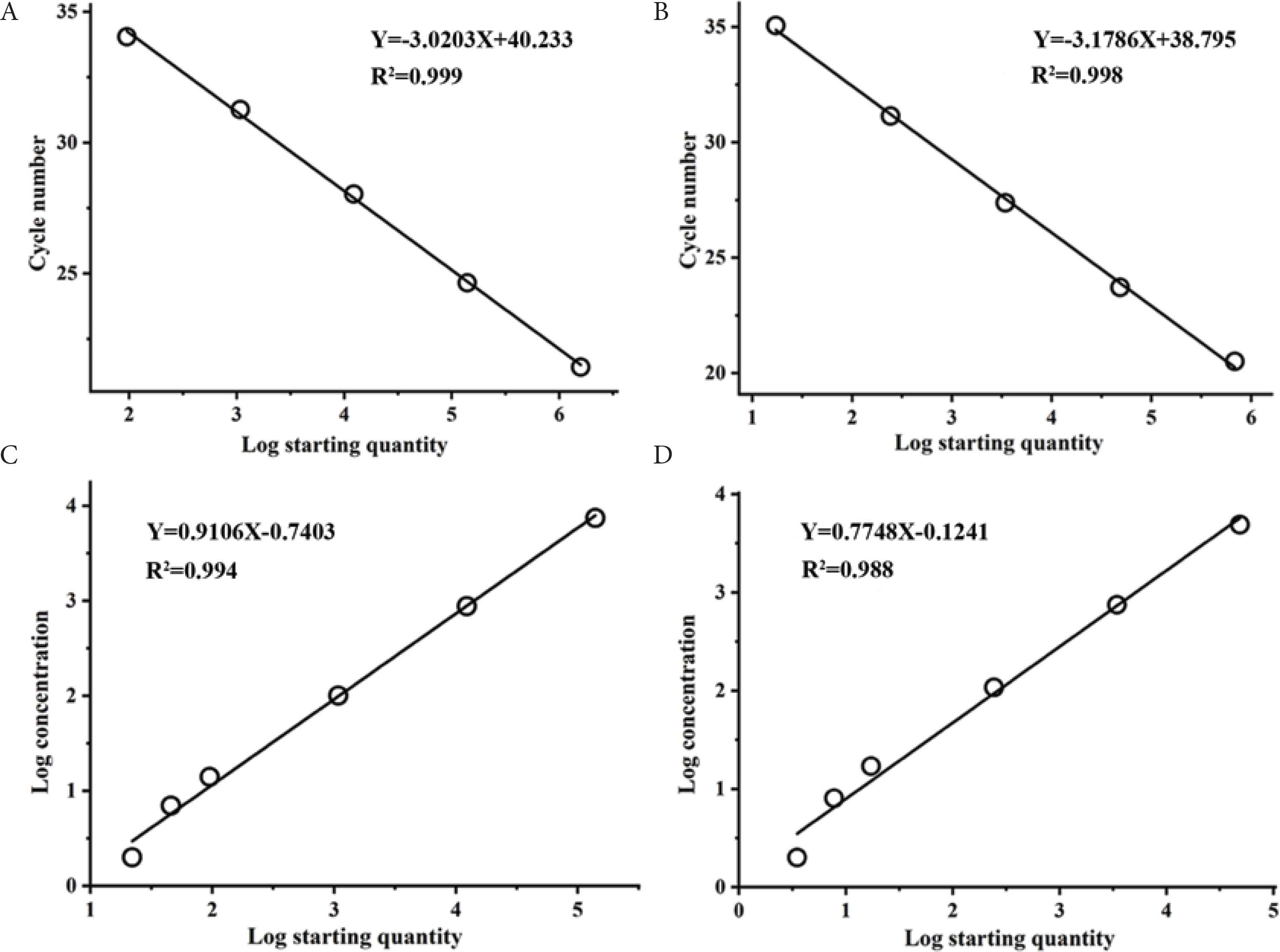
Quantification of serially diluted recombinant pUC57-APP and pUC57-HPS by ddPCR and qPCR. (A) and (B) Standard curves of qPCR were constructed using serial dilution of pUC57-APP and pUC57-HPS, respectively. The quantification correlation of qPCR was obtained by plotting the quantitative cycle value against the log starting concentration. (C) and (D) Standard curves of ddPCR were constructed using serial dilution of pUC57-APP and pUC57-HPS, respectively. The quantification relation of ddPCR between the log determined concentration and the log starting concentration.
3.4. Application and Detection of Clinical Samples
DNA extracted from 50 lung tissue of healthy pigs were confirmed negative by ELISA for A. pleuropneumoniae and H. parasuis and used as a negative control. For these negative samples, both qPCR and ddPCR were negative for A. pleuropneumoniae and H. parasuis. 12 A. pleuropneumoniae and 16 H. parasuis positive clinical samples were confirmed by ELISA and PCR, and both qPCR and ddPCR showed 100% positive and no false-positive results were shown. Another 112 were suspected A. pleuropneumoniae or H. parasuis infected samples. Among 112 clinical suspected samples, the positive detection rate for A. pleuropneumoniae by using qPCR and ddPCR was 7.1% and 10.7%, respectively; and the positive detection rate for H. parasuis was 8.9% in qPCR and 12.5% in ddPCR, respectively (Tables 4 and 5). All samples accurately detected by qPCR can also be detected by ddPCR, and the suspected clinical samples were further confirmed by ELISA. Compared with qPCR, ddPCR showed a higher positive detection rate, indicating that ddPCR assay is a more efficient method for the diagnose of A. pleuropneumoniae and H. parasuis suspected mixed infected clinical samples. The kappa value, which is used to measure agreement between two methods, was 0.781 and 0.814 for A. pleuropneumoniae and H. parasuis, indicating that qPCR and ddPCR assays have a good agreement.
| Detection methods | ddPCR | Total | |
|---|---|---|---|
| Positive | Negative | ||
| qPCR | 8 | 0 | 8 |
| 4 | 100 | 104 | |
| 12 | 100 | 112 | |
Results of ddPCR and qPCR assay for detection of A. pleuropneumoniae in clinical suspected samples
| Detection methods | ddPCR | Total | |
|---|---|---|---|
| Positive | Negative | ||
| qPCR | 10 | 0 | 10 |
| 4 | 98 | 102 | |
| 14 | 98 | 112 | |
Results of ddPCR and qPCR assay for detection of H. parasuis in clinical suspected samples
4. DISCUSSION
The surveillance of A. pleuropneumoniae and H. parasuis depends on the rapid and accuracy diagnosis. Conventional laboratory methods are less sensitive, laborious and time-consuming. Because of the precision and specificity of ddPCR, especially in the serial dilutions of genomic DNA from both pathogens, ddPCR is an effective method to detect bacterial DNA [28]. In addition, compared with other PCR methods, the difference of ddPCR is that each template molecule is amplified in an independent reaction chamber generated by water droplets emulsified in oil [26]. Each droplet acts as an independent micro-reactor, so it can achieve highly efficient amplification. Therefore, at the single-molecule resolution level, the sensitivity of ddPCR is higher than that of qPCR.
In this study, the serially diluted plasmid was used to measure the sensitivity of qPCR and ddPCR. As a novel molecular method, ddPCR has the advantage of absolute quantification detection of nucleic acids without relying on references or calibration curves. Our duplex ddPCR assay was specific and more sensitive than qPCR in detecting A. pleuropneumoniae and H. parasuis simultaneously. The detection of clinical samples that were identified by ELISA confirms that ddPCR has more accurate sensitivity. However, ddPCR is sometimes not as universal as qPCR due to the multiple partitions generated in the test. Furthermore, ddPCR may provide non-linear results when the initial concentration of the sample is too high. As described in the instruction manual of ddPCR, it is not practical for the detection of high concentration with more than 105 copies of pathogens in clinical samples. Therefore, qPCR could be used when the pathogen concentration is very high (>105 copies in the added template), while ddPCR is acceptable for the clinical samples with a low concentration (<105 copies/μL for A. pleuropneumoniae and 45 copies/μL for H. parasuis in the added template). Nevertheless, the cost of ddPCR reagents is comparatively high compared with qPCR for conventional diagnostic work, which may restrict its use and makes it hard to spread.
In the scope of our knowledge, this is the first assay for sensitive and simultaneous identification of A. pleuropneumoniae and H. parasuis by qPCR and ddPCR. In related molecular detection studies, limits of detection of A. pleuropneumoniae and H. parasuis were reported. A qPCR assay for detecting H. parasuis showed a sensitivity of LoD was 9.5–0.83 CFU per reaction for the boiling method of DNA extraction and 47.5–0.42 CFU per reaction for the PrepMan Ultra method; and for detecting A. pleuropneumoniae performed an analytical sensitivity of five colony forming units/reaction [18,29]. A loop-mediated isothermal amplification assay for detecting H. parasuis displayed a sensitivity of LoD was 0.2 pg/μL and for detecting A. pleuropneumoniae performed at a sensitivity of 8 CFU per tube [30,31]. A multiplex PCR assay for detecting A. pleuropneumoniae, Pasteurella multocida and H. parasuis showed the minimum detection concentration of 100 DNA copies, 10 DNA copies and 7 DNA copies, respectively [32]. In this study, the LoD (2 copies) by duplex ddPCR for both pathogens was lower than the published results. ddPCR may be useful to detect low concentration of clinical tissue samples of A. pleuropneumoniae and H. parasuis.
5. CONCLUSION
We established a duplex ddPCR assay for A. pleuropneumoniae and H. parasuis with higher specificity and greater sensitivity to those of qPCR. The duplex ddPCR assay is accurate for detecting both pathogens in serial dilutions. What’s more, the applicability of ddPCR is evaluated in clinical samples, which exhibits a higher positive detection rate than qPCR. The established method in this study can be a new approach to simultaneous quantitative detection of A. pleuropneumoniae and H. parasuis, so as to reduce the spread of swine diseases and to certain extent ensure the safety of meat food.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
WW contributed in literature search, experimental data collection and analyses, manuscript writing and editing. LS contributed in conceptualization and project administration. XC contributed in visualization, methodology, supervision, manuscript review.
ACKNOWLEDGMENTS
This work was supported by
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - Wen Wen AU - Lei Shi AU - Xun Chen PY - 2020 DA - 2020/09/25 TI - Simultaneous Detection of Actinobacillus pleuropneumoniae and Haemophilus parasuis in Pig by Duplex Droplet Digital PCR JO - eFood SP - 369 EP - 376 VL - 1 IS - 5 SN - 2666-3066 UR - https://doi.org/10.2991/efood.k.200918.001 DO - 10.2991/efood.k.200918.001 ID - Wen2020 ER -