
Identification of Putative Cell-entry-inhibitory Peptides against SARS-CoV-2 from Edible Insects: An in silico Study
- DOI
- 10.2991/efood.k.200918.002How to use a DOI?
- Keywords
- COVID-19; edible insect; gastrointestinal digestion; peptide; spike protein
- Abstract
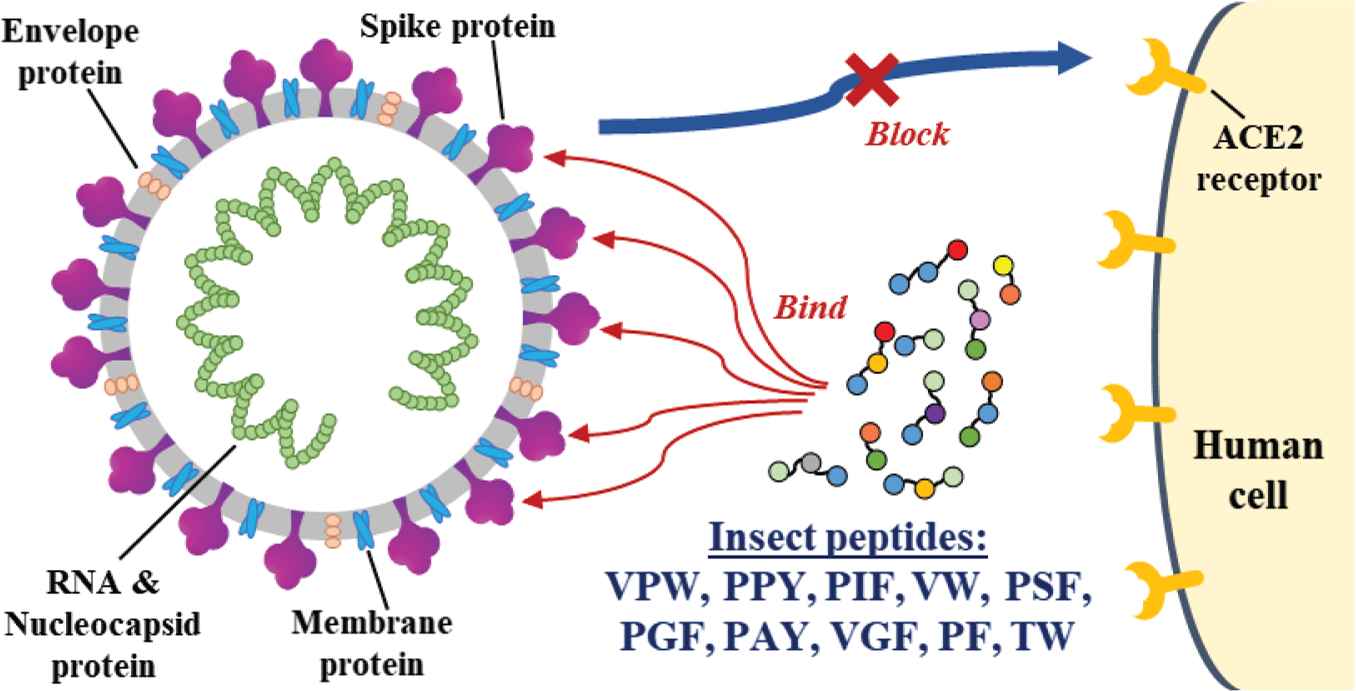
Coronavirus Disease 2019 (COVID-19), an infectious disease caused by SARS-CoV-2, has become the most contagious pandemic in the world. The binding of SARS-CoV-2 spike glycoprotein Receptor-binding Domain (RBD) to human angiotensin-converting enzyme 2 receptor precedes cell entry. Thus, RBD of SARS-CoV-2 spike glycoprotein is a key target for the development of drugs and vaccines to curb COVID-19. The aim of this in silico study was to identify potential SARS-CoV-2 cell entry inhibitors from peptides derived from edible insects. Twenty-four major proteins from mealworms, silkworm cocoons and housefly larvae were subjected to in silico gastrointestinal (GI) digestion, yielding 3560 fragments. Further screening led to 82 high-GI-absorption peptides with unique sequences. Molecular docking revealed 10 promising peptides (VPW, PPY, PIF, VW, PSF, PGF, PAY, VGF, PF and TW), predicted to interact with at least one key binding residue on RBD. Notably, VPW had the lowest docking energy score (−144.359) and binding affinity (−7.0 kcal/mol), highlighting its potency among the ten peptides. Binding affinities of the insect peptides were superior to some reported natural products. Our study suggests that when consumed, edible insects may be a source of putative SARS-CoV-2 cell entry inhibitors in the form of RBD-binding peptides.
- Graphical Abstract
- Copyright
- © 2020 The Authors. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
The current outbreak of Coronavirus Disease 2019 (COVID-19), caused by the newly discovered Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), has driven the world to a public health emergency. Globally, as of 5 August 2020, 18,354,342 confirmed cases of COVID-19, including 696,147 deaths, have been reported to the World Health Organization (https://covid19.who.int). People infected by SARS-CoV-2 might suffer from severe respiratory illness, along with headache, runny nose and fever; severe cases of COVID-19 may even lead to multi-organ failure and deaths [1]. SARS-CoV-2 is a single-stranded, positive sense RNA virus with four major structural proteins, namely the spike, membrane, envelope and nucleocapsid proteins [2]. The binding between the Receptor-binding Domain (RBD) of the spike glycoproteins and the Human Angiotensin Converting Enzyme 2 (hACE2) facilitate viral attachment onto human cells, which precedes membrane fusion and the subsequent SARS-CoV-2 infection [1,3,4]. Receptor recognition is a critical determinant of the entry of SARS-CoV-2 into the human host cells [5]. Thus, blocking the binding of RBD of SARS-CoV-2 spike glycoprotein to hACE2 could be a promising strategy toward the prevention and/or treatment of COVID-19 [4,6].
In recent years, molecular docking has become an increasingly popular drug discovery tool on account of its quick, inexpensive and accurate prediction of biological activity and interactions of ligand compounds with target molecules [7,8]. With this approach, information on the bond types, binding affinity and interactions between two molecules can be predicted from the best-fit conformation [9]. With the availability of the crystal structure information of SARS-CoV-2-hACE2 complex, there has been an increasing number of molecular docking studies that screen and identify natural and synthetic inhibitors as well as repurposed drugs that could target the interactions between the SARS-CoV-2 and hACE2 [1,2,8,10]. To the best of our knowledge, in contrast to the studies on phytochemicals, few molecular docking studies have investigated the potential of food-derived peptides in blocking the SARS-CoV-2–hACE2 interactions.
Food-derived peptides are known to exhibit diverse bioactivities, including antiviral properties [11]. Furthermore, peptides are often the preferred choice of therapeutic agents over conventional drugs because of their higher specificity, efficiency, and biodegradability, as well as lower molecular weight and toxicity [12]. In this regard, food-derived peptides which could serve as host cell entry inhibitors against SARS-CoV-2 are of specific interest in this study. Specifically, we are interested in peptides that are released from edible insects upon gastrointestinal (GI) digestion in the human body. Edible insects are good sources of bioactive compounds with great potential for pharmaceutical uses [13]. In this study, the mealworm, silkworm cocoon and housefly larvae were investigated. The three insect samples were chosen because previous studies [14–16] have highlighted their nutritional and medicinal values. The aim of this study was to identify potential SARS-CoV-2 cell entry inhibitors from edible insects through in silico analysis, focusing on high GI-absorption peptides that could bind to the RBD of the SARS-CoV-2 spike proteins. The outcome of this research could contribute towards the future development of food-derived peptides, specifically edible insect peptides, into therapeutic agents that target SARS-CoV-2.
2. MATERIALS AND METHODS
2.1. Materials
Figure 1 shows a flowchart of the study. A total of 24 major proteins found in three edible insects, namely mealworms, housefly larvae and silkworm cocoons [14–16], were studied. Sequences of the proteins were downloaded from the UniProt web server (http://www.uniprot.org) [17] on 14 July 2020.
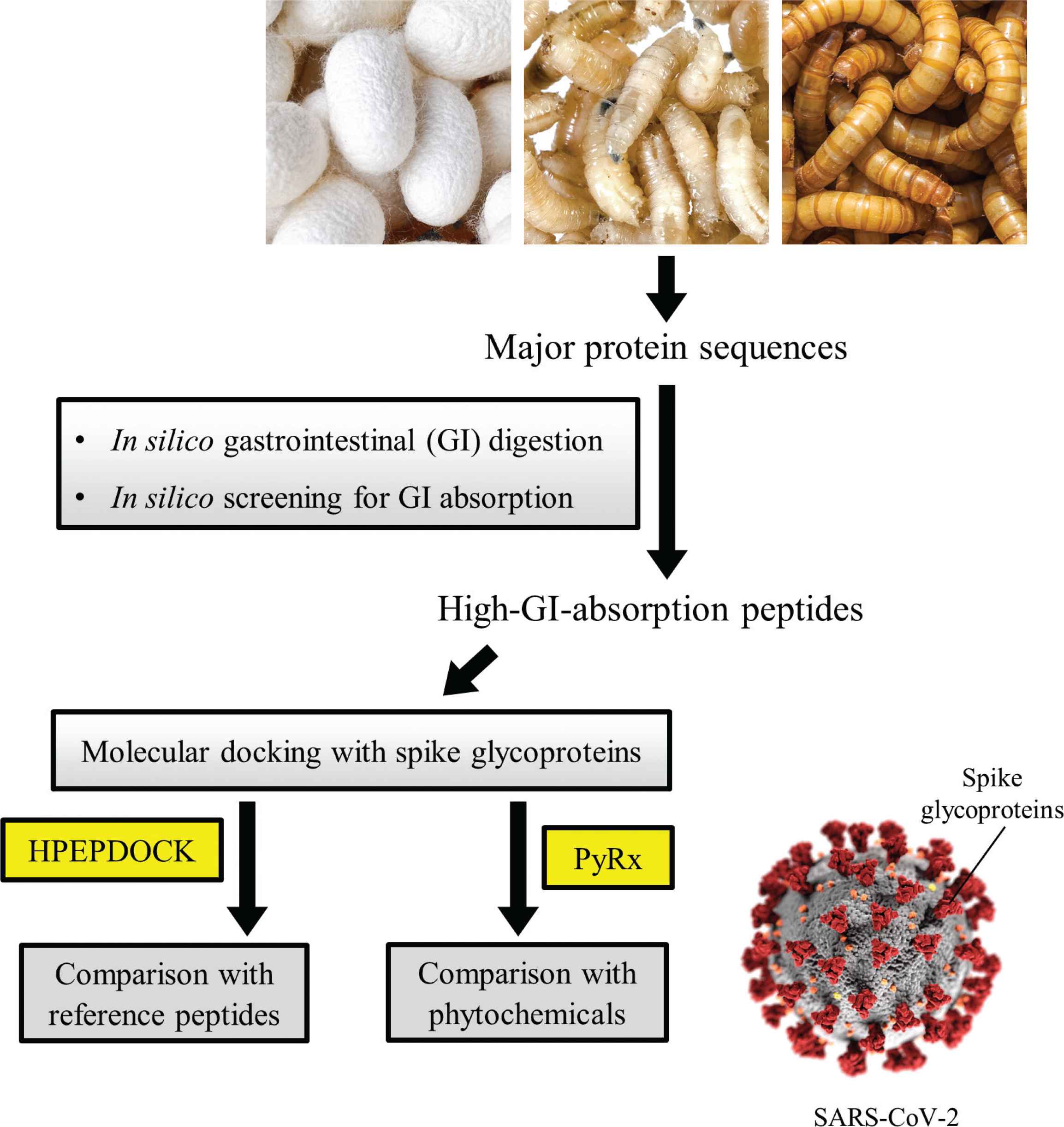
An overview of the study (SARS-CoV-2 photo credit: CDC/ Alissa Eckert, MSMI; Dan Higgins, MAMS).
2.2. In silico GI Digestion and Screening for GI Absorption
In silico GI digestion was performed by using the BIOPEP-UWM web server [18] as previously described [19,20]. The aforementioned 24 protein sequences were subjected to hydrolysis by pepsin (EC 3.4.23.1), trypsin (EC 3.4.21.4) and chymotrypsin A (EC 3.4.21.1) to simulate in silico GI digestion. The number of residues and the distribution of peptide length among the resultant peptide fragments were recorded. Next, unique peptide sequences comprising at least two amino acid residues were converted into Simplified Molecular-Input Line-Entry System (SMILES) strings by using the online tool PepSMI (https://www.novoprolabs.com/tools/convert-peptide-to-smiles-string). Using the SMILES strings generated as input, the potential GI absorption of the GI digestion-released peptide fragments was analyzed by using SwissADME (http://www.swissadme.ch). SwissADME is a free bioinformatics web tool that can be used to evaluate the penetration of molecules through the GI wall [21]. Following the screening step, peptide fragments predicted to have high GI absorption were taken to the following step of molecular docking analysis.
2.3. Molecular Docking Analyses with HPEPDOCK
The crystal structure of SARS-CoV-2 Spike RBD in complex with the hACE2 (PDB ID: 6LZG) [3] was obtained from the Protein Data Bank (http://www.rcsb.org/pdb) [22,23]. Next, the preparation of A chain (hACE2) and B chain (RBD) were performed by using BIOVIA Discovery Studio Visualizer (BIOVIA, Dassault Systèmes, BIOVIA Discovery Studio Visualizer, Version 20.1.0.192, San Diego: Dassault Systèmes, 2020), adding in polar hydrogens, and removing water, ligands and heteroatoms. Both structures were then saved in the PDB format and used for analysis on the HPEPDOCK server [24]. The prepared B chain was uploaded as receptor input, whereas the prepared A chain was uploaded as binding site reference. High-GI-absorption peptides released from our in silico GI digestion and four reference peptides that were previously reported to bind to the SARS-CoV-2 Spike RBD were entered in the FASTA format as peptide input. The resultant docking energy scores of peptide samples were tabulated. Interactions of the peptides with the SARS-CoV-2 Spike RBD in the docked models were visualized by using the LigPlot+ v.2.2 [25,26]. The docked models were checked for interactions, especially hydrogen bond and hydrophobic interactions, between the peptides and RBD residues. Based on the best-score models computed by HPEPDOCK, the binding poses of selected insect peptides on the surface of the RBD of SARS-CoV-2 spike proteins were visualized by using the ProteinsPlus web service (https://proteins.plus) [27].
2.4. Molecular Docking Analyses with AutoDock Vina Tool in PyRx
Following HPEPDOCK analysis, high-GI-absorption peptides predicted to bind to at least one of the nine known key binding residues (Leu455, Phe456, Ser459, Gln474, Ala475, Phe486, Phe490, Gln493 and Pro499) on RBD [4] were selected for further comparison with 26 phytochemicals that were previously reported to bind to the SARS-CoV-2 Spike RBD. Molecular docking analysis of the selected insect peptides and reference phytochemicals was performed by using the AutoDock Vina option in PyRx 0.8 [28,29]. For macromolecule preparation, the RBD protein was pre-processed by using the AutoDockTools 1.5.6 [30,31], which involved the removal of water as well as addition of Kollman charges and polar hydrogens, and then saved in the PDBQT format file. For the ligand preparation, the 3D structures of reference phytochemicals, where available, were retrieved from PubChem (https://pubchem.ncbi.nlm.nih.gov) [32] in the SDF format and converted into PDB format with the Open Babel tool in PyRx 0.8. Where only 2D information is found on PubChem for a reference phytochemical, the 2D-to-3D structure conversion was accomplished by using the MarvinSketch software (version 20.13.0) (ChemAxon, https://www.chemaxon.com), saved in the PDB format. The 3D structures of the selected insect peptides were obtained from the collection of 3D structures of dipeptides and tripeptides made available by Prasasty and Istyastono [33]. The docking process was performed in which the binding site of protein was set with the grid box center x-, y-, z-axis values of −25.0426, 15.8143, −25.4535 with dimension of 100 × 100 × 90 Å, encompassing 21 binding site residues (Lys417, Gly446, Tyr449, Tyr453, Leu455, Phe456, Tyr473, Ala475, Gly476, Glu484, Phe486, Asn487, Tyr489, Phe490, Gln493, Gly496, Gln498, Thr500, Asn501, Gly502 and Tyr505) [3] and three other RBD residues (Ser459, Gln474 and Pro499) that were reported to be important for RBD-hACE2 binding [4]. The resultant binding affinities of peptide samples and reference phytochemicals were tabulated.
3. RESULTS AND DISCUSSION
In order to discover insect peptides that are potentially bioavailable upon oral consumption, simulated GI digestion was performed on insect proteins in this in silico study. A total of 3560 peptide fragments were released from the 24 insect proteins analyzed following in silico GI digestion; 1771 peptide fragments were released from the silkworm cocoon, 1006 fragments from the mealworm, and 783 fragments from housefly larvae. Details of each insect protein and the outcome of in silico GI digestion are presented in Table 1. The distribution of peptide length in the GI digestion-released fragments varied among the three insect samples. When 2- and 3-residue peptide fragments generated by in silico GI digestion are considered collectively, the percentages of such peptides released from the proteins of the silkworm cocoon, housefly larvae, and mealworm are in the range of 25.53–57.14%, 28.89–45.45% and 27.78–43.08%, respectively. Low-molecular-weight peptides are absorbed more readily through the GI tract than the high-molecular-weight peptides. Most often, di- and tri-peptides can be absorbed intact through the epithelium of the GI tract [34]. In accordance with this, the greatest number of high-GI-absorption peptides was predicted for the silkworm cocoon (172 fragments) by the SwissADME web server [21], in contrast with 140 high-GI-absorption peptides for mealworm and 105 for housefly larvae.
| Insect | Protein | UniProt accession | Number of fragments released | Distribution of peptide length (%) | Number of peptides with high GI absorption | ||||
|---|---|---|---|---|---|---|---|---|---|
| 2 residues | 3 residues | 4 residues | 5 residues | > 5 residues | |||||
| Mealworm | Alpha-actinin-4 | E0VM19 | 390 | 25.38 | 17.69 | 8.72 | 3.85 | 4.87 | 58 |
| Actin-like | S5M0Y7 | 132 | 23.48 | 18.18 | 9.09 | 4.55 | 13.64 | 25 | |
| Tropomyosin 2 | V5JDH8 | 107 | 25.23 | 13.08 | 9.35 | 4.67 | 10.28 | 13 | |
| Calponin | D2A180 | 65 | 20.00 | 13.85 | 3.08 | 16.92 | 9.23 | 8 | |
| Troponin T | D3TS62 | 144 | 26.39 | 15.97 | 5.56 | 6.25 | 5.56 | 16 | |
| 28-kDa desiccation stress protein | Q27013 | 80 | 22.50 | 15.00 | 2.50 | 5.00 | 17.50 | 11 | |
| 13-kDa hemolymph protein a | Q7YWD2 | 36 | 13.89 | 13.89 | 13.89 | 5.56 | 13.89 | 3 | |
| 12-kDa hemolymph protein b | Q7YWD7 | 52 | 25.00 | 13.46 | 3.85 | 9.62 | 5.77 | 6 | |
| Silkworm cocoon | Fibroin heavy chain | P05790 | 380 | 11.84 | 13.68 | 14.47 | 0.26 | 53.68 | 33 |
| Fibroin light chain | P21828 | 82 | 15.85 | 13.41 | 12.20 | 4.88 | 17.07 | 8 | |
| Fibrohexamerin | P04148 | 93 | 32.26 | 16.13 | 8.60 | 4.30 | 4.30 | 17 | |
| Sericin 1 | P07856 | 275 | 20.36 | 13.09 | 8.00 | 4.36 | 24.36 | 28 | |
| Sericin 2 | D2WL76 | 652 | 38.50 | 7.98 | 16.26 | 8.44 | 5.06 | 74 | |
| Sericin 3 | A8CEQ1 | 227 | 13.66 | 14.54 | 3.08 | 11.01 | 40.09 | 1 | |
| Seroin 1 | H9IZK4 | 21 | 33.33 | 23.81 | 9.52 | 14.29 | 9.52 | 4 | |
| Seroin 2 | Q8T7L7 | 41 | 26.83 | 14.63 | 9.76 | 4.88 | 9.76 | 7 | |
| Housefly larvae | Ferritin | T1PLJ3 | 85 | 11.76 | 25.88 | 8.24 | 1.18 | 8.24 | 6 |
| Cytochrome c | T1PF88 | 45 | 20.00 | 8.89 | 13.33 | 4.44 | 6.67 | 6 | |
| Catalase | T1PCG9 | 197 | 22.34 | 13.20 | 10.66 | 5.08 | 7.61 | 27 | |
| Peroxiredoxin | T1PEX1 | 82 | 26.83 | 8.54 | 7.32 | 12.20 | 8.54 | 12 | |
| Antifungal peptide-1 | G9B2K0 | 72 | 27.78 | 11.11 | 0.00 | 9.72 | 12.50 | 14 | |
| Superoxide dismutase | G3GJ67 | 92 | 21.74 | 13.04 | 9.78 | 3.26 | 7.61 | 12 | |
| Superoxide dismutase [Cu-Zn] | Q6SCL6 | 44 | 34.09 | 11.36 | 11.36 | 2.27 | 18.18 | 8 | |
| Glutathione reductase family member | P91884 | 166 | 22.29 | 11.45 | 13.86 | 8.43 | 11.45 | 20 | |
Outcome of in silico GI digestion of major insect proteins
More specifically, in silico GI digestion of the proteins of each insect released the largest number of peptide fragments from sericin 2 (silkworm cocoon; 652 fragments), alpha-actinin-4 (mealworm; 390 fragments), and catalase (housefly larvae; 197 fragments). Unsurprisingly, the three insect proteins were also the key contributors of high-GI-absorption peptides, as predicted by the SwissADME web server [21], in the descending order of sericin 2 (74 fragments) > alpha-actinin-4 (58 fragments) > catalase (27 fragments). Notably, the combination of sericin 2 and fibroin heavy chain contributed to 76% of the 172 high-GI-absorption peptides generated from silkworm cocoon major proteins upon in silico GI digestion. Taken together, our in silico analysis suggests that silkworm cocoon proteins, when consumed, are more likely to produce GI-absorbable peptides which could be bioavailable in vivo. Specifically, the sericin 2 and fibroin heavy chain of the silkworm cocoon deserve future attention in the search of natural, edible sources of bioavailable peptides.
Since the aim of this study was to identify putative SARS-CoV-2 cell entry inhibitors from edible insect peptides that are potentially bioavailable, we analyzed only the 82 high-GI-absorption insect peptides in our molecular docking studies. In HPEPDOCK analysis, all the high-GI-absorption peptides were docked to the spike glycoprotein RBD via the local peptide docking mode, taking hACE2 as the reference for binding sites. The docking energy scores of the peptides ranged from −53.873 to −144.359, as documented in Table 2. Recently, Yang et al. [35] reported a strong linear correlation between the in vitro activity of monoamine oxidase A inhibitory peptides and the HPEPDOCK docking energy scores predicted for the peptides when docked to the enzyme. Their finding highlights the possible application of HPEPDOCK scores for ranking or comparing predictively the relative bioactivity of peptides. In this study, based on docking energy scores, the five top-score peptides of mealworms in descending order are VPW, PPY, PW, IW, and PAY. For silkworm cocoon, the five top-score peptides in descending order are PIF, VW, IPY, PGF and TW. In the case of housefly larvae, the five top-score peptides in descending order are VW, PSF, PGF, VGF, and PF. The same high-GI-absorption peptide sequences might be released from different proteins of same insect, or from proteins of different insects. Thus it is unsurprising that VW and PGF were both listed in the five top-score peptides of silkworm cocoon and housefly larvae. Importantly, VPW was the peptide with the lowest (most negative) docking energy score among the whole set of 82 high-GI-absorption insect peptides, implying its relative potency in binding with SARS-CoV-2 spike protein RBD.
| Insect peptides/Reference peptides | Docking energy scores | Sources | ||
|---|---|---|---|---|
| Mealworm | Silkworm cocoon | Housefly larvae | ||
| IEEQAKTFLDKFNHEAEDLFYQS* | −223.534 | - | - | - |
| FLDKFNHEAEDLFYQSSL* | −216.761 | - | - | - |
| VPW | −144.359 | √ | - | - |
| LPIY* | −139.134 | - | - | - |
| CSNAIPEL* | −135.411 | - | - | - |
| PPY | −131.773 | √ | - | - |
| PIF | −131.740 | - | √ | - |
| PW | −128.975 | √ | - | - |
| VW | −127.336 | - | √ | √ |
| PSF | −126.842 | - | - | √ |
| IW | −125.121 | √ | - | - |
| IPY | −124.618 | - | √ | - |
| PGF | −124.263 | - | √ | √ |
| PAY | −124.041 | √ | - | - |
| VGW | −122.073 | √ | - | - |
| VGF | −117.686 | - | - | √ |
| PF | −117.215 | - | - | √ |
| TW | −116.374 | - | √ | - |
| GPY | −116.337 | √ | √ | - |
| AW | −114.853 | √ | √ | - |
| PY | −113.683 | √ | - | √ |
| GW | −112.521 | - | √ | √ |
| GPF | −111.835 | - | √ | - |
| AVF | −109.368 | √ | - | - |
| AGF | −108.132 | √ | √ | - |
| VY | −106.424 | - | √ | - |
| PH | −106.229 | √ | - | √ |
| IY | −105.689 | - | √ | - |
| TF | −103.645 | √ | √ | √ |
| PIL | −102.956 | - | - | √ |
| IF | −102.463 | √ | √ | √ |
| TY | −102.385 | √ | - | - |
| PPK | −101.851 | √ | - | - |
| GY | −100.116 | √ | √ | √ |
| QF | −99.749 | √ | √ | - |
| AY | −99.248 | √ | √ | √ |
| CF | −99.034 | √ | √ | - |
| EF | −98.684 | √ | √ | - |
| SF | −98.372 | √ | √ | √ |
| AF | −97.583 | √ | √ | √ |
| VF | −97.546 | - | √ | √ |
| GF | −97.025 | - | - | √ |
| PGL | −95.628 | - | - | √ |
| PVL | −95.210 | √ | - | - |
| IIL | −93.084 | √ | - | - |
| VIL | −92.851 | √ | - | |
| IH | −92.003 | √ | - | √ |
| DF | −91.415 | √ | √ | √ |
| AH | −86.858 | - | - | √ |
| PM | −88.607 | - | - | √ |
| AIL | −85.500 | √ | - | - |
| IGV | −84.507 | - | √ | - |
| VH | −83.877 | - | √ | √ |
| IM | −83.596 | √ | - | - |
| PL | −81.722 | √ | √ | √ |
| VVL | −81.706 | - | - | √ |
| GVK | −81.566 | √ | - | - |
| IGL | −81.030 | - | - | √ |
| VPL | −80.901 | - | - | √ |
| GIL | −80.882 | √ | √ | - |
| GH | −80.350 | - | √ | - |
| IAL | −80.321 | - | √ | - |
| VM | −79.125 | - | √ | √ |
| PK | −78.120 | √ | √ | √ |
| VAL | −75.726 | √ | √ | - |
| AAL | −75.648 | - | √ | √ |
| IK | −74.883 | √ | √ | √ |
| GAL | −73.325 | - | - | √ |
| VGL | −73.179 | - | - | √ |
| EL | −71.453 | √ | - | √ |
| AM | −71.421 | - | - | √ |
| IL | −70.938 | √ | - | √ |
| GGL | −70.675 | - | √ | - |
| AGL | −69.661 | - | - | √ |
| GM | −69.515 | √ | - | √ |
| TL | −68.382 | √ | √ | √ |
| VL | −63.197 | √ | √ | √ |
| VK | −62.584 | √ | √ | √ |
| GK | −61.557 | √ | √ | √ |
| AL | −61.453 | √ | √ | √ |
| CL | −59.264 | √ | √ | √ |
| SL | −58.303 | √ | √ | √ |
| GL | −57.232 | √ | - | √ |
| EV | −56.956 | √ | - | - |
| DL | −56.133 | √ | √ | √ |
| AK | −53.873 | √ | √ | √ |
Indicates reference peptides.
Docking energy scores of the top peptide binding models generated from the high-GI-absorption insect peptides and the SARS-CoV-2 spike protein RBD
In this study, we also compared the docking energy scores of the high-GI-absorption insect peptides with reference peptides previously reported or predicted to bind to SARS-CoV-2 RBD, namely IEEQAKTFLDKFNHEAEDLFYQS, FLDKFNHEAEDLFYQSSL, LPIY and CSNAIPEL. IEEQAKTFLDKFNHEAEDLFYQS, also named as SBP1, is a 23-mer peptide designed from the peptidase domain of hACE2 [36]. FLDKFNHEAEDLFYQSSL is an artificially-designed 18-residue peptide [37]. LPIY and CSNAIPEL are peptides identified from the myosin of scallop Mizuhopecten yessoensis after in silico enzymatic hydrolysis [38]. The docking energy scores of IEEQAKTFLDKFNHEAEDLFYQS and FLDKFNHEAEDLFYQSSL are both lower than that computated for all high-GI-absorption insect peptides (Table 2). On the other hand, the docking energy scores of LPIY and CSNAIPEL are both slightly higher than VPW, although still lower than the rest of the insect peptides. Notably, although the four reference peptides are either comparable or superior to the insect peptides in docking energy scores, all four were predicted to be susceptible to in silico GI digestion by BIOPEP server and to have low GI absorption by SwissADME server (data not shown). In short, where oral bioavailability is concerned, the insect peptides presented in Table 2 would still have an advantage over the four reference peptides. In particular, the mealworm-derived VPW is the most promising peptide in our high-GI-absorption insect peptide dataset to play the role of SARS-CoV-2 cell entry inhibitor.
Five insect peptides having the lowest docking energy scores were chosen from each insect sample and further analyzed by using LigPlot+. Figure 2 shows the binding poses of the insect peptides on the RBD surface. Table 3 summarizes the RBD residues that interact with each peptide, in addition to the types and the number of interactions, as computed by LigPlot+. Figures 3 and 4 present the graphic representations that reveal the molecular interactions at the reference peptide- and insect peptide-RBD interfaces, respectively. According to Wang et al. [3] and Yi et al. [4], 24 residues on RBD are known to bind to hACE2. In addition, Yi et al. [4] reported that the nine residues Leu455, Phe456, Ser459, Gln474, Ala475, Phe486, Phe490, Gln493 and Pro499 are the key binding sites on RBD; binding to these residues can significantly affect the affinity of RBD for hACE2. Peptides that are able to interact with any of the nine key binding residues would be potential inhibitors that block the binding of the RBD on spike glycoprotein to hACE2, hence preventing virus entry into the host cell and the initiation of infection [4]. In our LigPlot+ analysis, the two reference peptides IEEQAKTFLDKFNHEAEDLFYQS and FLDKFNHEAEDLFYQSSL were found to interact with more key binding residues (Leu455, Phe456, Ala475 and Gln493) than were the insect peptides or the other two reference peptides LPIY and CSNAIPEL. Moreover, in comparison with the other peptides, IEEQAKTFLDKFNHEAEDLFYQS and FLDKFNHEAEDLFYQSSL also interact with a greater number of other RBD binding site residues, mainly via hydrophobic interactions. The two reference peptides each establish at least 15 hydrophobic interactions with the RBD structure, surpassing all other peptides we analysed (Table 3 and Figure 3). This observation substantiates the much lower docking energy scores calculated by HPEPDOCK for the two peptides, relative to the rest of the peptides analysed (Table 2). Further supporting the importance of hydrophobic interactions at the RBD-peptide interface is our observation that despite the lack of hydrogen bond formation, the lowest docking energy score was predicted for the RBD-VPW docked model relative to the rest of the insect peptides. All other insect peptides form at least one hydrogen bond with a key binding site residue and/or other binding site residues as revealed by our LigPlot+ analysis (Table 3).
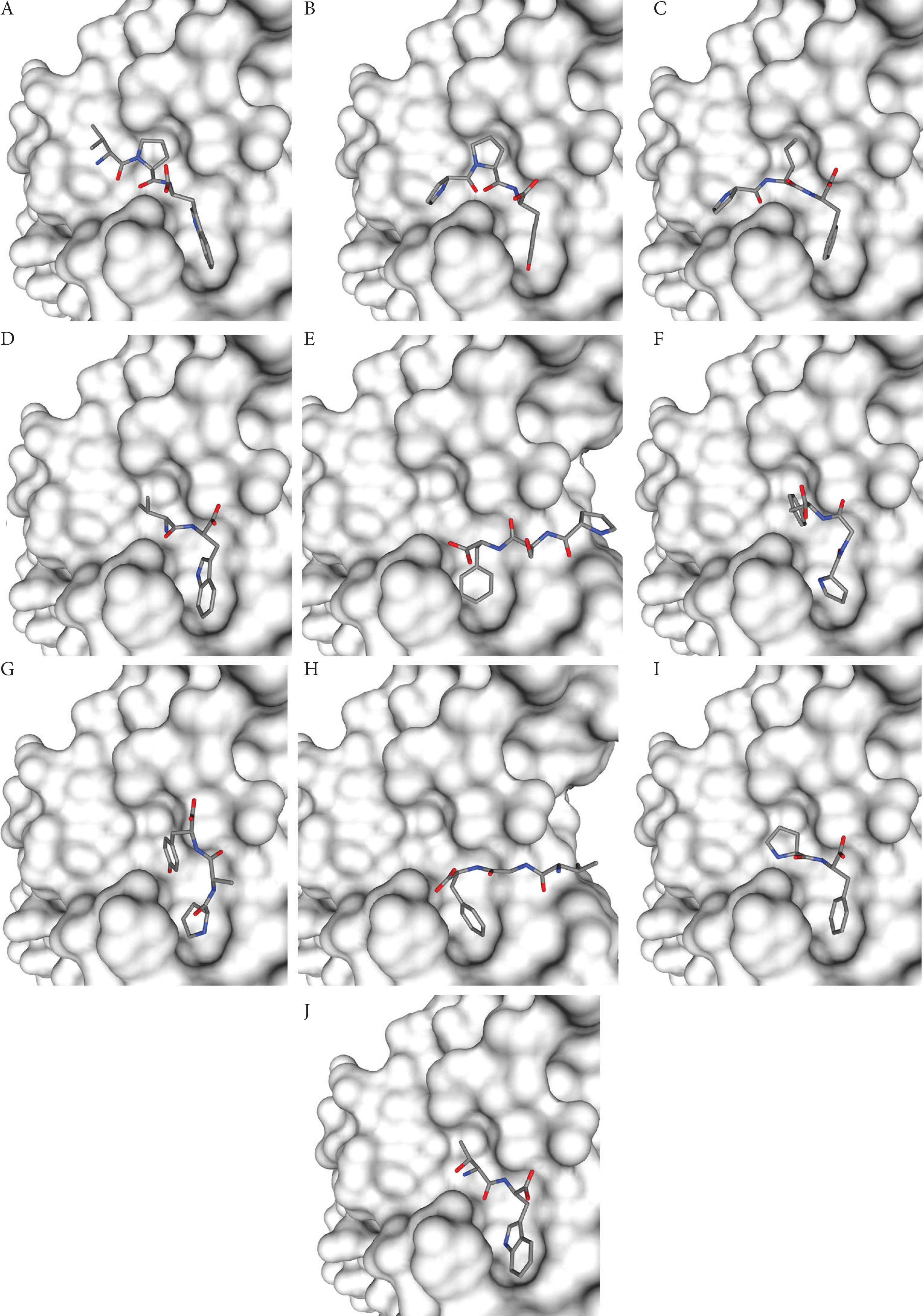
Binding poses of 10 selected insect peptides on the surface of the RBD of SARS-CoV-2 spike protein. (A) VPW, (B) PPY, (C) PIF, (D) VW, (E) PSF, (F) PGF, (G) PAY, (H) VGF, (I) PF, and (J) TW.
| Insect peptides/Reference peptides | Sources | Interactions with RBD residues# | Total number of interactions | ||||
|---|---|---|---|---|---|---|---|
| Mealworm | Silkworm cocoon | Housefly larvae | Hydrogen bond | Hydrophobic interaction | Hydrogen bond | Hydrophobic interaction | |
| IEEQAKTFLDKFNHEAEDLFYQS* | - | - | - | Tyr453, Gln493, Asn501 | Lys417, Tyr453, Leu455, Phe456, Tyr473, Ala475, Asn487, Tyr489, Gln493, Ser494, Gln498, Thr500, Asn501, Gly502, Tyr505 | 4 | 15 |
| FLDKFNHEAEDLFYQSSL* | - | - | - | Tyr489, Tyr505 | Arg403, Lys417, Tyr449, Tyr453, Leu455, Phe456, Ala475, Asn487, Tyr489, Gln493, Ser494, Tyr495, Gly496, Phe497, Gln498, Asn501, Tyr505 | 2 | 17 |
| VPW | √ | - | - | - | Arg403, Glu406, Gln409, Gly416, Lys417, Tyr453, Leu455, Tyr495, Gly496, Phe497, Asn501, Tyr505 | 0 | 12 |
| LPIY* | - | - | - | Glu406, Tyr453, Gln493, Gly496 | Arg403, Glu406, Tyr449, Tyr453, Gln493, Ser494, Tyr495, Gly496, Gln498, Asn501, Gly502, Tyr505 | 4 | 12 |
| CSNAIPEL* | - | - | - | Arg403, Tyr453, Thr500, Asn501, Gly502 | Arg403, Lys417, Tyr449, Tyr453, Leu455, Ser494, Tyr495, Gly496, Thr500, Asn501, Tyr505 | 6 | 11 |
| PPY | √ | - | - | Arg403, Tyr453, Tyr505 | Arg403, Asp405, Glu406, Lys417, Tyr453, Leu455, Tyr495, Phe497, Tyr505 | 4 | 9 |
| PIF | - | √ | - | Tyr453 | Arg403, Asp405, Glu406, Lys417, Tyr453, Leu455, Tyr495, Phe497, Tyr505 | 1 | 9 |
| PW | √ | - | - | Arg403, Tyr453 | Arg403, Glu406, Lys417, Tyr453, Ser494, Tyr495, Gly496, Phe497, Asn501, Tyr505 | 2 | 10 |
| VW | - | √ | √ | Arg403, Glu406, Tyr453, Gln493 | Arg403, Glu406, Tyr453, Gln493, Ser494, Tyr495, Gly496, Asn501, Tyr505 | 5 | 9 |
| PSF | - | - | √ | Arg403, Gln493 | Arg403, Tyr449, Tyr453, Gln493, Ser494, Tyr495, Gly496, Phe497, Tyr505 | 2 | 9 |
| IW | √ | - | - | Tyr505 | Arg403, Tyr453, Tyr495, Gly496, Phe497, Asn501, Tyr505 | 1 | 7 |
| IPY | - | √ | - | Arg403, Tyr453, Gly496, Tyr505 | Arg403, Glu406, Arg408, Gln409, Lys417, Tyr453, Tyr495, Gly496, Phe497, Tyr505 | 4 | 10 |
| PGF | - | √ | √ | Tyr453 | Arg403, Glu406, Tyr453, Gln493, Ser494, Tyr495, Gly496, Phe497, Asn501, Tyr505 | 1 | 10 |
| PAY | √ | - | - | Glu406, Tyr453, Gln493 | Arg403, Glu406, Tyr453, Leu455, Gln493, Tyr495, Gly496, Phe497, Asn501, Tyr505 | 3 | 10 |
| VGF | - | - | √ | Arg403, Tyr453 | Arg403, Tyr449, Tyr453, Gln493, Ser494, Tyr495, Gly496, Phe497, Asn501, Tyr505 | 2 | 10 |
| PF | - | - | √ | Tyr453 | Arg403, Glu406, Lys417, Tyr453, Gln493, Ser494, Tyr495, Gly496, Asn501, Tyr505 | 2 | 10 |
| TW | - | √ | - | Arg403, Glu406, Tyr453, Gln493 | Arg403, Glu406, Tyr453, Ser494, Tyr495, Gly496, Asn501, Tyr505 | 4 | 8 |
Residues in bold are among the nine known key binding residues on RBD that hACE2 binds to, whereas the underlined residues are other binding sites for hACE2.
Indicates reference peptides.
Interactions between the SARS-CoV-2 RBD residues and five peptides having the lowest docking energy scores from each insect sample
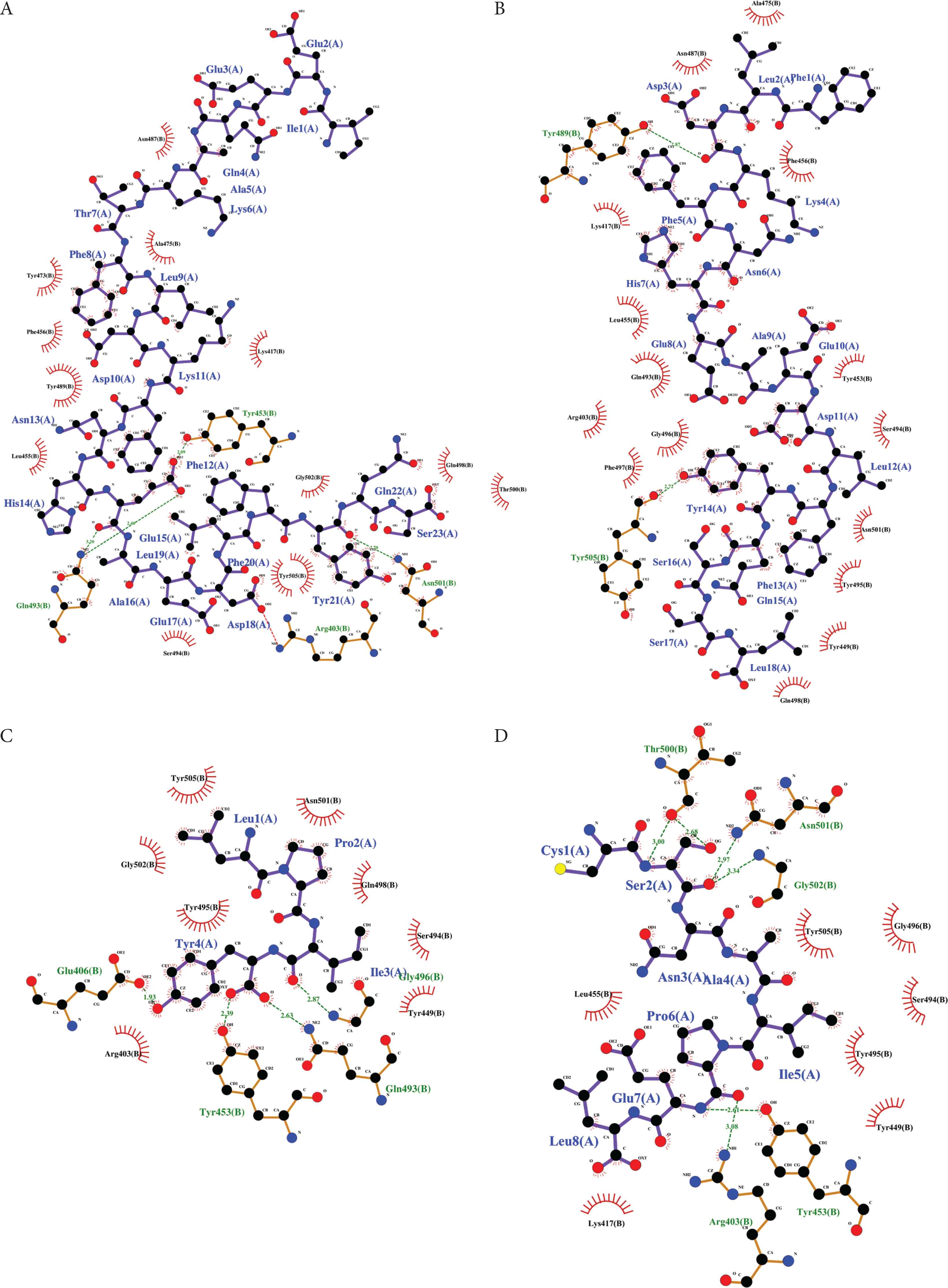
Two dimensional visualization of the RBD-peptide binding interfaces as computed by LigPlot+ for the reference peptides (A) IEEQAKTFLDKFNHEAEDLFYQS, (B) FLDKFNHEAEDLFYQSSL, (C) LPIY and (D) CSNAIPEL. Ligands (peptides) are in purple, whereas the bonds of protein (RBD of SARS-CoV-2 spike protein) are represented in orange color. Hydrogen bonds are displayed in green dashed lines, whereas hydrophobic interactions are represented by red spoked arcs.
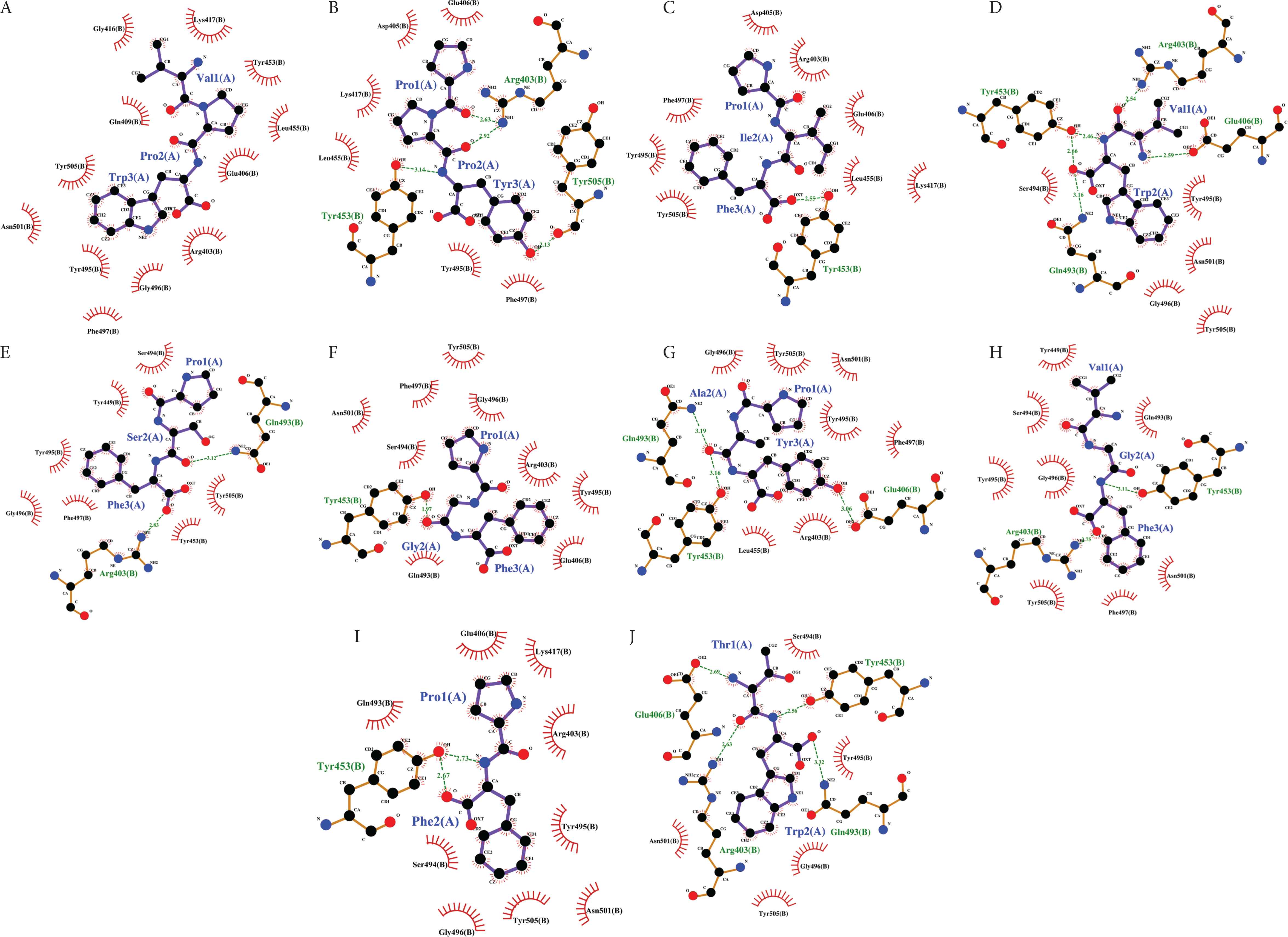
Two dimensional visualization of the RBD-peptide binding interfaces as computed by LigPlot+ for the insect peptides: (A) VPW, (B) PPY, (C) PIF, (D) VW, (E) PSF, (F) PGF, (G) PAY, (H) VGF, (I) PF, and (J) TW. Descriptions for ligands (peptides), protein (RBD of SARS-CoV-2 spike protein), and bonds are the same as in Figure 3.
In this study, VPW was predicted to interact with the SARS-CoV-2 spike protein through 12 hydrophobic interactions, which involve one key binding residue Leu455 and five other residues previously reported to bind to hACE2 (Lys417, Tyr453, Gly496, Asn501, Tyr505) among others (Table 3 and Figure 4A). This clearly indicates the potential of VPW to bind to the RBD of SARS-CoV-2 spike proteins and to block its interaction with the human cell surface receptor hACE2. PPY and PIF are the peptides computed by HPEPDOCK to have the second and the third lowest docking energy scores. The two peptides each form nine hydrophobic interactions at the RBD-peptide interface, similarly interacting with one key binding residue Leu455 and three other binding site residues (Lys417, Tyr453 and Tyr505). The slightly lower docking energy score of PPY, relative to PIF, is apparently attributable to PPY forming four hydrogen bonds with the RBD of the spike protein, whereas PIF forms only one (Figures 4B and 4C). LPIY can form both hydrophobic interaction and hydrogen bond with the key binding residue Gln493. We also observed the same for insect peptides VW, PSF and PAY (Figures 4D, 4E and 4G). In addition, like CSNAIPEL, insect peptides VPW, PPY, PIF, and PAY are each able to form hydrophobic interaction with the key binding residue Leu455 (Figures 4A–4C and 4G). Thus Leu455 and Gln493 are two of the nine key binding residues targeted by the insect peptides in our dataset.
At present, the number of studies concerning natural or food-derived peptides which potentially bind to SARS-CoV-2 spike proteins is limited. Hence we decided to further characterise the insect peptides by comparing their RBD-binding affinities with those of phytochemicals. Comparison was made with 26 natural products derived from edible and medicinal plants that were reported in 10 studies to have the ability to bind to SARS-CoV-2 spike proteins; such natural products represent potential cell entry inhibitors against SARS-CoV-2 [2,10,39–46]. To this end, we focussed on 10 insect peptides (VPW, PPY, PIF, VW, PSF, PGF, PAY, VGF, PF and TW) which were predicted by LigPlot+ to bind to at least one key binding residue on the RBD of SARS-CoV-2 spike proteins. Molecular docking analysis by using the PyRx software (AutoDock Vina option) found that among the insect peptides, VPW displayed the lowest binding affinity value (−7.0 kcal/mol), suggesting that it can bind more strongly to the SARS-CoV-2 spike proteins than the other nine insect peptides (Figure 5). In contrast with the docking energy scores computed by HPEPDOCK, the difference of binding affinities between peptides as analyzed by PyRx was less distinct, for example, between PIF, VW and PSF. This may be attributed to the greater accuracy of HPEPDOCK in analyzing small peptides when compared with traditional small-molecule docking programs, including AutoDock Vina [47]. All in all, the trends of docking energy scores and binding affinities generally appear similar for the 10 insect peptides, both suggesting the strongest effectiveness of VPW in binding to the SARS-CoV-2 spike protein. When compared with the phytochemicals, the binding affinity of VPW was found to be 4.11–17.65% weaker than digitoxin, hesperidin, glycyrrhizic acid, taraxerol and asparoside-C. The binding affinity of VPW is comparable to those of dihydrotanshinone I, fisetin, friedelin and asparoside-D. By contrast, the binding affinity of VPW was 2.86–42.86% stronger than 17 of the phytochemicals analyzed. The binding affinities of nine natural products, namely curcumin, carvacrol, thymol, cinnamyl acetate, l-4-terpineol, pulegone, cinnamaldehyde, anethole and geraniol, were weaker than the 10 insect peptides. Taken together, the 10 insect peptides in Figure 5 are comparable or superior to some phytochemicals previously reported to have binding potential toward the SARS-CoV-2 spike proteins. This suggest that like the natural products of edible and medicinal plants, insect peptides could be a natural resource of SARS-CoV-2 cell entry inhibitors which deserves more research attention.

Binding affinity of ten insect peptides in comparison with selected phytochemicals. Bars represent mean ± standard errors (n = 3). Black bars represent insect peptides. EGCG-3Me, epigallocatechin 3-O-(3-O-methyl) gallate.
4. CONCLUSION
Ten peptides were identified from the in silico GI digestion of major proteins from edible insects, which were predicted in silico to be readily absorbed by the GI tract. Importantly, the peptides could potentially serve as cell entry inhibitors against SARS-CoV-2. Molecular docking suggests mealworm-derived peptide VPW is the most promising among the ten peptides. Our study highlights that edible insects, especially mealworms, are promising sources of potential anti-COVID-19 peptides, which should be explored more intensively in future. Importantly, future in vivo experiments are necessary to confirm our findings; despite high GI absorption, in vivo bioavailability could also be influenced by alteration or inactivation by the liver.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
FCW and TTC contributed in conceptualization. JHO and TTC contributed in literature search, experimental data collection and analyses. FCW and TTC contributed in study design. TTC and JHO contributed in manuscript writing and editing.
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - Fai-Chu Wong AU - Joe-Hui Ong AU - Tsun-Thai Chai PY - 2020 DA - 2020/09/25 TI - Identification of Putative Cell-entry-inhibitory Peptides against SARS-CoV-2 from Edible Insects: An in silico Study JO - eFood SP - 357 EP - 368 VL - 1 IS - 5 SN - 2666-3066 UR - https://doi.org/10.2991/efood.k.200918.002 DO - 10.2991/efood.k.200918.002 ID - Wong2020 ER -