
Plasma Cell Leukemia – Facts and Controversies: More Questions than Answers?
- DOI
- 10.2991/chi.k.200706.002How to use a DOI?
- Keywords
- Plasma cell leukemia; plasma cell dyscrasia; chemotherapy; novel agents; stem cell transplantation; clinical trials; prognostic index
- Abstract
Plasma cell leukemia (PCL) is an aggressive hematological malignancy characterized by an uncontrolled clonal proliferation of plasma cells (PCs) in the bone marrow and peripheral blood. PCL has been defined by an absolute number of circulating PCs exceeding 2.0 × 109/L and/or >20% PCs in the total leucocyte count. It is classified as primary PCL, which develops de novo, and secondary PCL, occurring at the late and advanced stages of multiple myeloma (MM). Primary and secondary PCL are clinically and biologically two distinct entities. After the diagnosis, treatment should be immediate and should include a proteasome inhibitor and immunomodulator-based combination regimens as induction, followed by stem cell transplantation (SCT) in transplant-eligible individuals who have cleared the peripheral blood of circulating PCs. Due to the rarity of the condition, there have been very few clinical trials. Furthermore, virtually all of the myeloma trials exclude patients with active PCL. The evaluation of response has been defined by the International Myeloma Working Group and consists of both acute leukemia and MM criteria. With conventional chemotherapy, the prognosis of primary PCL has been ominous, with reported overall survival (OS) ranging from 6.8 to 12.6 months. The use of novel agents and autologous SCT appears to be associated with deeper response and an improved survival, although it still remains low. The PCL prognostic index provides a simple score to risk-stratify PCL. The prognosis of secondary PCL is extremely poor, with OS of only 1 month.
- Copyright
- © 2020 International Academy for Clinical Hematology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
Plasma cell leukemia (PCL) is an aggressive plasma cell dyscrasia characterized by an uncontrolled clonal proliferation of plasma cells (PCs) in the bone marrow (Figure 1) and peripheral blood (Figure 2) [1]. The first case was reported by Gluziński and Reichenstein more than a century ago [2]. The definition of PCL has traditionally been based on Kyle’s 1974 criteria [3]. Present diagnostic criteria include absolute number of circulating PCs exceeding 2.0 × 109/L and/or >20% PCs in the total leucocyte count [4] (Figure 2).
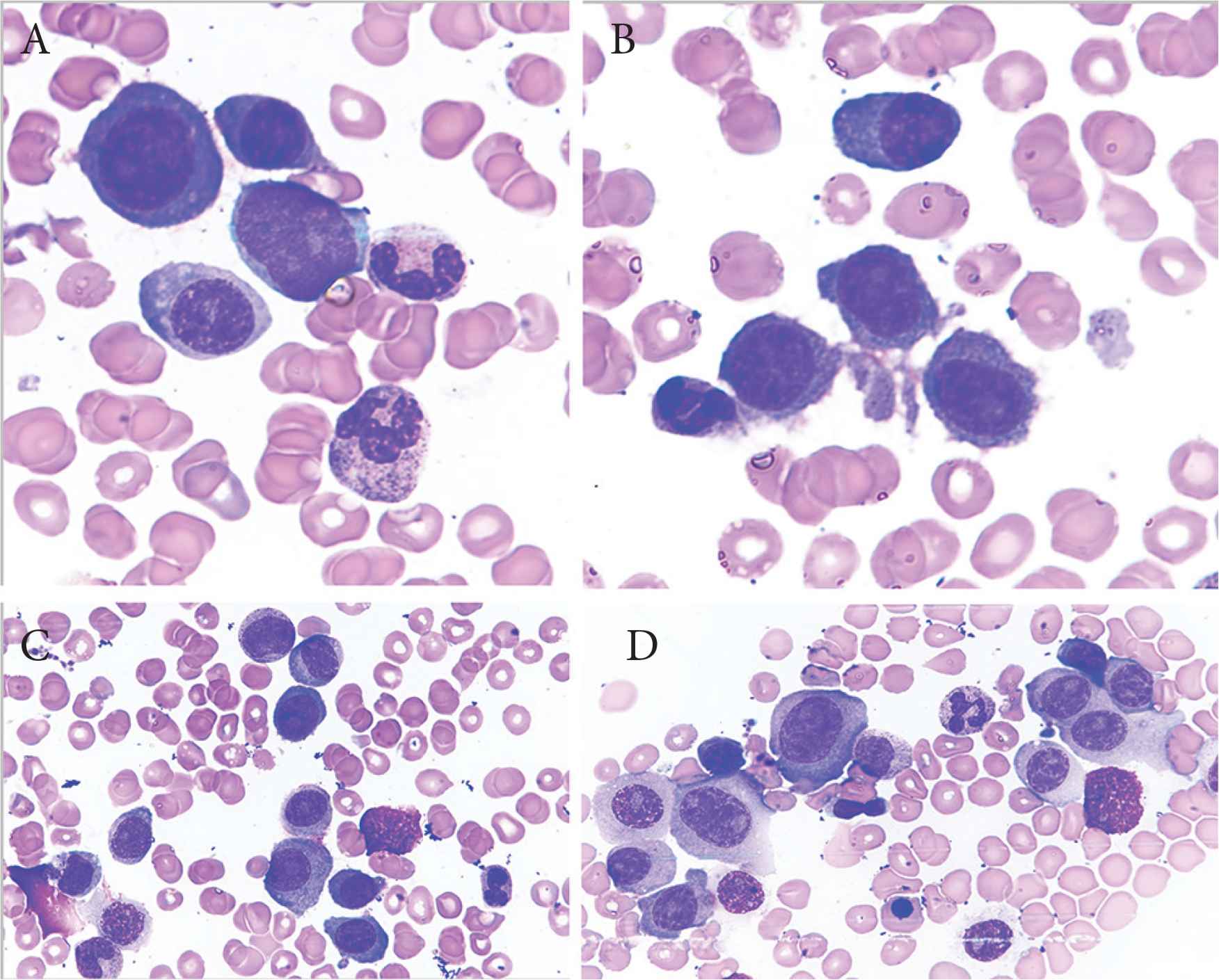
Bone marrow aspirate smears showing immature plasma cells including plasmablasts (A–C) and atypical binucleate plasma cell (D). Obj. magn. 1000×, Wright’s staining; from the Department of Hematology, the University Hospital in Krakow.
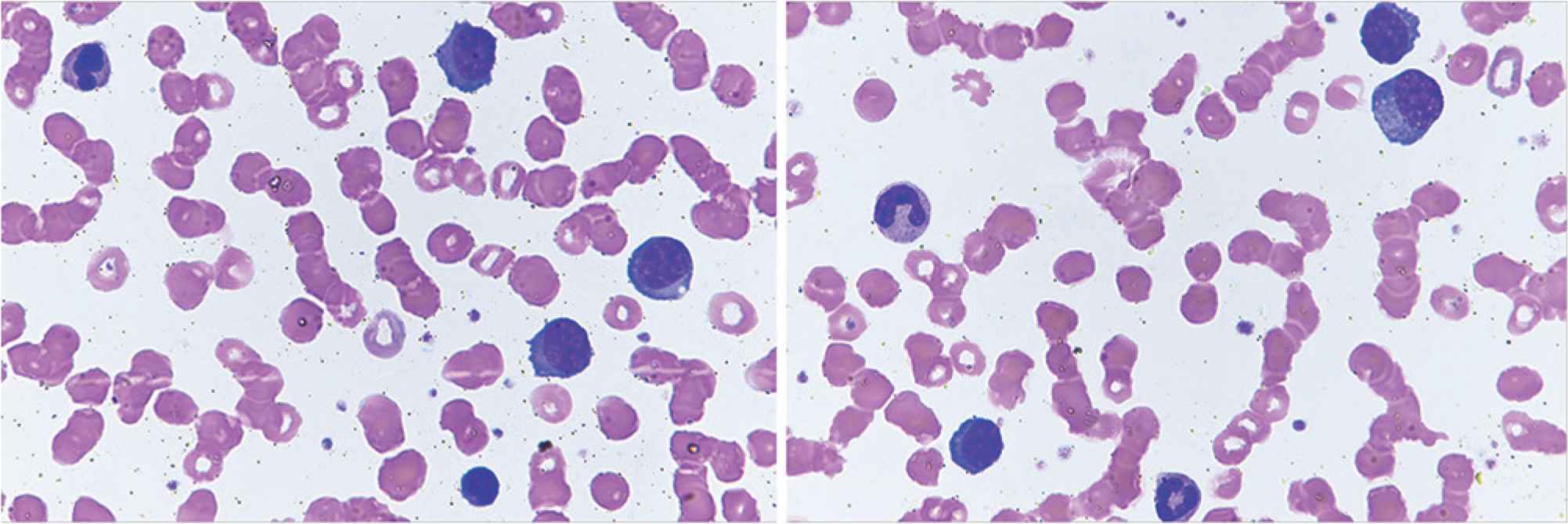
Peripheral blood smears showing plasma cells and red cells rouleaux formation. Obj. magn. 1000×, Wright’s staining; from the Department of Hematology, the University Hospital in Krakow.
In general, PCs from patients with PCL overlap in antigenic expression with those from patients with multiple myeloma (MM). The unique biology of PCL results from the disruption of the mechanisms crucial for homing of malignant plasma cells within the bone marrow [4]. A number of adhesion molecules and chemokine receptors are involved in this process. The most important findings in PCL include lower expression of neural cell adhesion molecule (CD56) and leukocyte function-associated antigen-1, which normally enable to anchor PCs to the bone marrow stroma [5,6], increased secretion of metalloproteinase-9 leading to the excessive degradation of all components of the extracellular matrix, and as a consequence, to a weaker myeloma cell interaction [7,8], and high expression of very late antigen-4 (VLA-4) (integrin α4β1) that favors invasiveness of leukemic cells by causing their extravasation in the way of contact with its ligand in capillary vessel wall [9,10]. All these changes result in the regression of PCs to the peripheral blood, making PCL a highly aggressive disease with extremely poor prognosis.
The incidence of PCL in Europe is evaluated at the level of 0.04 cases per 100,000 persons per year [11]. PCL is classified as primary (pPCL) when it develops de novo, and secondary (sPCL) when it occurs in patients with previously recognized MM, typically at a late and advanced stage of the disease [4]. Historically, pPCL has been reported as more common than sPCL, with their relative incidence estimated at 60–70% and 30–40%, respectively [12]. However, in recent years there has been an upward trend in sPCL, now accounting for about 50% of the cases [13], probably due to improved overall survival (OS) in MM patients. Notably, pPCL and sPCL are two clinically and biologically distinct entities which only share the features of plasma cells circulating in the peripheral blood, unfavorable course and prognosis.
The current knowledge on PCL is somewhat controversial, from the definition of the disease to the treatment algorithms and the evaluation of treatment outcomes. This review article underlines the most important issues, presenting various points of views. Because of the relative low incidence and prevalence of PCL, most data concerning clinical features, treatment approaches and results come from case reports and retrospective series [12,14–17].
2. CONTROVERSY OVER THE DIAGNOSIS
The current diagnostic criteria which are very restrictive have not been prospectively evaluated to determine a need for any modification. However, such an arbitrary approach might underestimate the real clinical significance of circulating PCs. In the era of next generation flow cytometry the definition of PCL is still under debate [18,19].
Conventional microscopic analysis of the peripheral blood sample should be performed in all MM patients who present with clinical symptoms suspicious of PCL. If there are more than 20% circulating PCs and/or an absolute PC count exceeding 2.0 × 109/L, the diagnosis of PCL should be established, according to current criteria [4]. Of note, patients with relapsed/refractory heavily pretreated MM, and with poor bone marrow reserve commonly have baseline leukopenia and may not develop significant absolute PCs but may meet percentage criteria. Therefore, there are some cases in the literature in which only one criterion was considered sufficient to establish a diagnosis and start treatment [13,20,21].
Recent studies have shown that even lower percentages of PCs in peripheral blood may be related to an adverse prognosis in newly diagnosed MM patients, reflecting the need for re-definition of the diagnostic cut-off [19,22–24]. As it has been proven, the presence of ≥5% circulating PCs in patients with MM has similar adverse prognostic impact as PCL defined traditionally, so that this level may be proposed as a new cut-off point [24]. Interestingly, Rupin et al. in their small retrospective study stated that, irrespective of quantity, the presence of any PC in the peripheral blood is a poor prognostic indicator [22]. Taking into consideration all the issues mentioned above, the International Myeloma Working Group (IMWG) is working on a new definition of PCL.
It is also important to state that the presence of a significant number of PCs in the peripheral blood, but polyclonal and not at high percentage as in PCL, can be transiently observed in non-malignant conditions, such as severe sepsis, mononucleosis and serum sickness. Thus, peripheral blood flow cytometry is a useful tool to verify the clonality of the PCs, and it should be a high priority in further studies, with a special emphasis on those conducted in newly diagnosed patients.
3. PRIMARY PLASMA CELL LEUKEMIA
3.1. Clinical and Biological Characteristics
Because of the more aggressive course of the disease, including a higher tumor burden and higher proliferative index at diagnosis, pPCL patients may present with symptoms of hypercalcemia, profound anemia or bleeding diathesis due to thrombocytopenia. Physical examination may reveal hepatomegaly, splenomegaly, enlarged lymph nodes, pleural effusion, neurologic deficits and palpable extramedullary plasmacytomas in the soft tissues [4]. Trephine biopsy often demonstrates extensive bone marrow PC infiltration, with anaplastic or plasmablastic morphology, resulting in a reduced bone marrow reserve. In the blood, laboratory tests demonstrate leukocytosis, elevated lactate dehydrogenase (LDH) and β2-microglobulin serum levels, as well as low levels of albumin. Except from PCL cases presenting with high proliferative rate, elevated LDH and very aggressive course, there are also cases with packed marrow and overflow from the marrow into the peripheral blood which are proved to be less aggressive.
Primary PCL is also often diagnosed as light chain or non-secretory disease. It presents with cytogenetic abnormalities and molecular findings, which are usually found only in advanced MM. On flow cytometry, PCs more often express CD20, CD44, CD45, CD19 and CD23, while CD27, CD56, CD71, CD117 and human leukocyte antigen DR (HLA-DR) are less frequently detected [25,26]. CD38, known as a target for immunotherapy in MM, is universally expressed in pPCL. Cytogenetic abnormalities are similar to those characteristics for MM, but occur with higher frequency. Notably, the proportions of certain genetic changes have different profile from that in MM. Based on the results of the last prospective trials [27,28], hyperdiploidy is very rare, while hypodiploidy, del13q, del17p, gain/loss of chromosome 1 and translocations involving immunoglobulin heavy chain locus are more frequently observed. The 14q32 translocation is common in pPCL, occurring in 87% cases. Analysis of rearrangements of the 14q32 region in pPCL cases compared with newly diagnosed MM patients stage III showed higher incidence of t(11;14) (33% vs. 16%; p < 0.025) and of t(14;16) (13% vs. 1%; p < 0.002), though the incidence of t(4;14) was identical in both groups [29]. More recent data from the comprehensive molecular analysis of a prospective series with 23 pPCL patients revealed that immunoglobulin heavy chain locus translocations were identified in 87% of cases, with a prevalence of t(11;14) and t(14;16) at the level of 40% and 30.5%, respectively [30].
Relapsed pPCL routinely presents with the same pattern as in the initial stage of the disease, including clinical features and natural history (rapid course, high tumor burden, high proliferative index, high leukocytosis, high LDH serum level, extramedullary involvement, bone marrow infiltration by leukemic PCs) [4].
3.2. Therapeutic Options – Still Not Enough
There are no precise guidelines for the treatment of pPCL. The recommendations concerning therapeutic approaches in PCL are supported by limited data and based mainly on expert opinion. Virtually all the MM trials exclude patients with active PCL. So far, no randomized, phase III trials have been conducted in pPCL, while only two prospective, phase II studies have been published [27,28]. Notably, after the diagnosis of PCL, immediate treatment should be initiated. The main aim is maximal cytoreduction.
3.2.1. Conventional chemotherapy
Following the IMWG consensus statement from 2013 [4], intensive multidrug traditional chemotherapy with alkylating agents or anthracyclines [such as hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone (HyperCVAD) and cisplatin, doxorubicin, cyclophosphamide, etoposide (PACE)] is considered to be effective. However, in terms of survival, (VAD)-based regimens, compared with combinations containing only an alkylating agent plus a corticosteroid, appear to have limited benefit [15,31,32].
3.2.2. New agents
Immunomodulatory drugs (IMIDs) and proteasome inhibitors (PIs), now widely used in antimyeloma therapy, have significantly improved survival of MM patients [33,34]. There is an increasing evidence in the literature that these agents may also improve the outcome in pPCL, but the benefit is less apparent when compared with MM. Bortezomib is probably the key new agent in pPCL, and, when used in drug combination, rapidly reduces tumor load and reverses complications such as renal failure and hypercalcemia [32]. In the 2018 European Myeloma Network (EMN) recommendations for the management of patients with pPCL, include bortezomib-based multidrug regimens in the frontline therapy [35] (Figure 3).
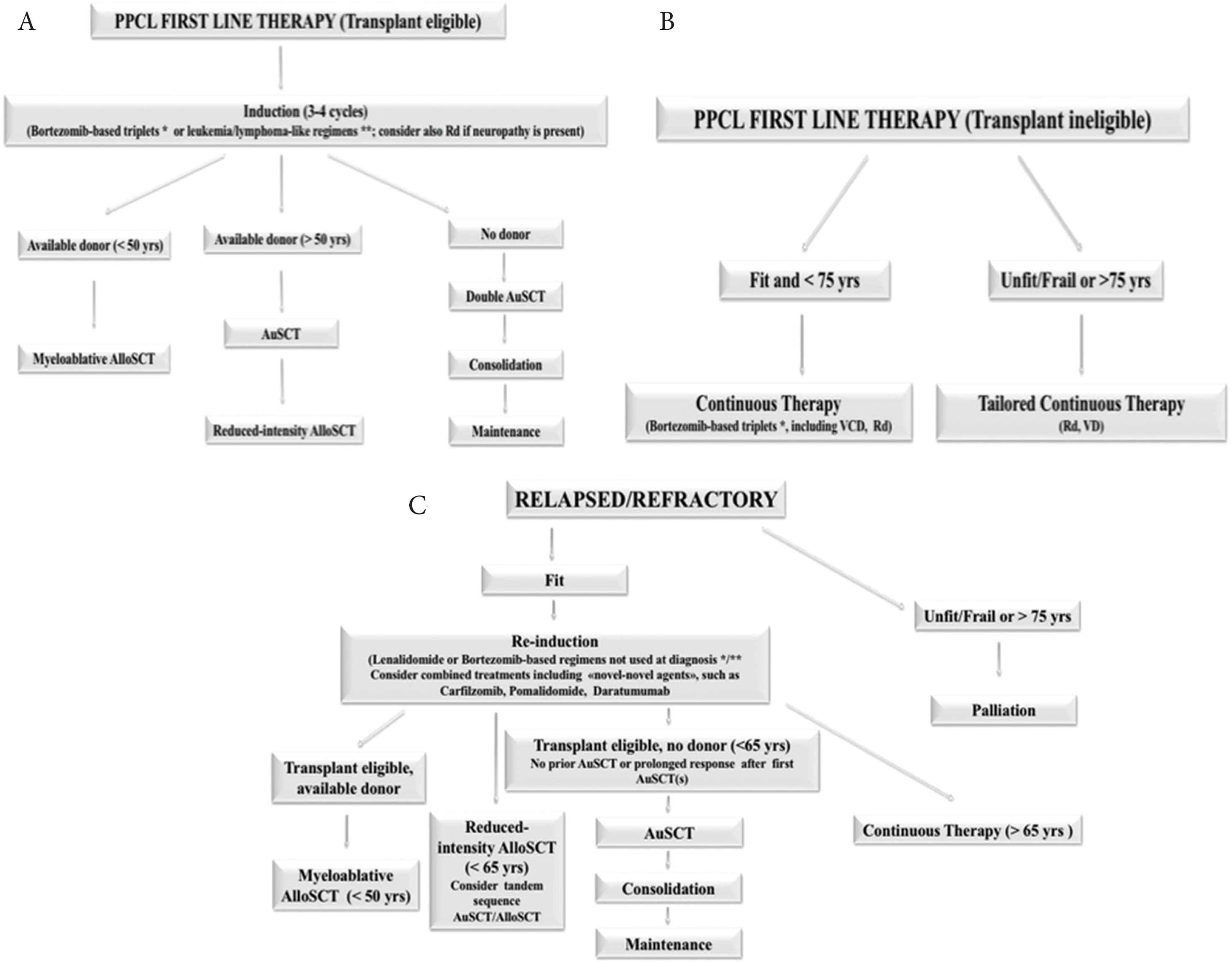
Therapeutic algorithm for primary plasma cell leukemia. The European Myeloma Network recommendations. (A) First line therapy in transplant eligible patients. (B) First line therapy for patients not eligible to transplantation. (C) Treatment for relapsed/resistant patients. Adapted from: Gavriatopoulou et al. [35]. AlloSCT, allogeneic stem cell transplantation; ASCT, autologous stem cell transplantation; Hyper-CVAD, hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone; PAD, bortezomib, doxorubicin, dexamethasone; Rd, lenalidomide, low-dose dexamethasone; VD, bortezomib and dexamethasone; VCD, bortezomib, cyclophosphamide, dexamethasone; *VRD, bortezomib, lenalidomide, dexamethasone; VTD, bortezomib, thalidomide, dexamethasone; VTD/VRD-PACE, bortezomib, thalidomide, lenalidomide, dexamethasone, continue infusion cisplatin, doxorubicin, cyclophosphamide, etoposide.
The preferred combination for fit individuals includes bortezomib-based triplets: bortezomib, thalidomide, and dexamethasone (VTD) or bortezomib, doxorubicin and dexamethasone (PAD). Young patients with high tumor burden (hyperleucocytosis and rapidly progressive disease), requiring a fast response, may benefit from aggressive chemotherapy such as hyperfractionated cyclophosphamide, vincristine, continue-infusion doxorubicin, bortezomib, dexamethasone or VTD/bortezomib, lenalidomide, dexamethasone (VRD)-PACE, but there is no clear evidence of its superiority [4,35].
In the European settings, induction therapy with bortezomib-based regimens, such as melphalan, prednisone, bortezomib (MPV), VRD or VTD, is the treatment of choice for transplant-ineligible individuals. In the USA, combined therapy with cyclophosphamide, such as bortezomib, cyclophosphamide, and dexamethasone (VCd), carfilzomib, cyclophosphamide, and dexamethasone (KCd), or quadruplets of bortezomib, cyclophosphamide, lenalidomide, and dexamethasone (VCRd) and carfilzomib, cyclophosphamide, lenalidomide, and dexamethasone (KCRd), and potentially daratumumab-based regimens are new options even in elderly patients.
3.2.3. Stem cell transplantation
Autologous stem cell transplantation (ASCT) may result in a subsequent outcome improvement in newly diagnosed pPCL, though without significant changes in treatment results in recurrent disease. In transplant-eligible patients younger than 65 years, a tandem autologous transplantation or tandem transplant with an ASCT followed by a reduced-intensity conditioning allogeneic transplantation are potential options [4].
Of note, transplant-eligible patients should avoid alkylating agents in the first-line therapy, in order to allow sufficient collection of CD34+ stem cells from the peripheral blood. ASCT following myeloablative treatment must be considered in all eligible patients who achieve a significant response after a course of three to four cycles of induction treatment [35]. Despite being less effective than in MM, high-dose melphalan followed by ASCT is currently the preferable conditioning regimen (200 mg/m2, with dose adjustment according to renal function) [36]. It has been reported that high-dose melphalan is safe even in elderly patients (≥70 years), while reduction of the conditioning dose to 140 mg/m2 results in inferior outcomes [37]. Some data also suggest a possible advantage of tandem-ASCT [38]; however, a recent retrospective European Society for Blood and Marrow Transplantation (EBMT) survey of has not confirmed the benefit [39].
There are no data comparing upfront allogeneic SCT (AlloSCT) to single or tandem ASCT. Although AlloSCT has a lower relapse rate, it is associated with a much higher risk of non-relapse-related mortality, without any survival advantage. Therefore, the advantages and disadvantages of frontline AlloSCT should be carefully assessed in each pPCL case [39].
3.2.4. Post-transplantation management
There is a strong rationale for consolidation and maintenance therapy with lenalidomide, in order to improve the depth of response, maintain remission and, possibly, to improve survival [40–42]. However, the use of PI, IMID or daratumumab-based regimens as maintenance is still under debate because of scarce data. Due to unequal access to drugs around the world, it should be stated that multidrug therapy (ideally including proteasome inhibitor) followed by ASCT and continuous maintenance is the preferred approach.
3.2.5. Emerging issues
So far, the treatment of transplant-ineligible elderly patients, as well as relapsed/refractory pPCL appears to be ineffective and disappointing [4,32,43]. Thus, those patients should be considered as candidates in clinical trials. In very old and/or frail individuals, personalized approach (i.e., dose- and time-adjusted drug combinations) should be implemented according to the tolerability, aiming to maintain patients on therapy for as long as possible. In case of relapse, a switch to drugs not previously used should be considered.
3.3. Ongoing Trials – Hope for the Future
While the t(11;14) translocation is detected in approximately 15–20% of MM patients, it is reported in up to 50% of pPCL cases. In some studies, it was reported as favorable, while in others it did not influence the prognosis [44,45]. So far, the t(11;14) appears to carry a standard risk [46]. It is a routinely investigated cytogenetic abnormality which, associated with high Bcl-2 expression, represents a useful tool for prediction of sensitivity to venetoclax. This highly selective Bcl-2 inhibitor was reported as a very promising drug in refractory pPCL when used as a part of combination treatment together with daratumumab, bortezomib and dexamethasone, enabling the induction of MRD negativity as assessed by 6-color flow cytometry with a 10−4 sensitivity [47]. Moreover, Jelinek et al. [48] provided indisputable evidence of venetoclax activity, as a single-agent, in the first relapse of a patient with pPCL with t(11;14), reflected in MRD negativity confirmed by both next generation flow (NGF) cytometry and positron emission tomography with 2-[18F]fluoro-2-deoxy-d-glucose integrated with computed tomography (FDG-PET/CT).
New therapeutic approaches for PCL will parallel those in MM, with next generation PIs/IMIDs, monoclonal antibodies, bispecific antibodies or chimeric antigen receptor T cells (CAR-T cells) [35].
Currently, there are a few ongoing trials available on ClinicalTrials.gov, mainly for patients with relapsed/refractory pPCL, including ixazomib (NCT02504359, NCT02547662), elotuzumab (NCT01729091), SVN53-67/M57-KLH (a peptide vaccine) (NCT02334865), panobinostat (NCT02506959) and umbilical-cord derived NK-cells in combination with elotuzumab, and lenalidomide, followed by ASCT (NCT01729091). In the European settings, the EMN is conducting a prospective, phase II trial (EMN-12/HOVON129, EudraCT Number 2016-003105-33) in younger and elderly newly diagnosed pPCL patients, with the use of carfilzomib and lenalidomide-based (KRd) treatment. The preliminary results on the group of 33 patients, 21 aged ≤65 years and 12 aged >65 years, were reported by van de Donk et al. [49]. KRd was shown as an effective combination for the rapid induction of deep hematologic response [≥very good partial response (VGPR) in 80% and ≥complete response (CR) in 33% after four cycles of therapy), without early deaths or treatment discontinuation because of toxicity.
The expression of CD38 is similar in PCL and MM [50]. As a highly and uniformly expressed antigen, it represents a target for the treatment with anti-CD38 monoclonal antibodies. Daratumumab is the most advanced anti-CD38 antibody in both development and wide use in the clinical practice. Although isatuximab, targeting a completely different epitope of the CD38 molecule, may also be promising. However, there is no ongoing clinical trial with anti-CD38 in PCL.
3.4. Response Criteria: One Disease, Dual Approach
Due to the leukemic nature of the disease, the increased incidence of light-chain only (Bence Jones) type and the high frequency of oligo/non-secretory forms, the evaluation of response in pPCL should follow the criteria for both acute leukemia and MM [51,52]. The role of rapid decrease of PCs in the peripheral blood and bone marrow infiltration has not been assessed, so far. However, the complete clearance of PCs from the peripheral blood, accompanied by bone marrow plasma cells drop below 5% are required to classify the response as complete remission. To exclude extramedullary involvement, imaging techniques (including FDG-PET/CT and MRI) should be implemented in the algorithm of the disease evaluation. According to the IMWG consensus statement, there are four categories of response (Table 1). All PCL patients require a careful evaluation of extramedullary disease at the moment of diagnosis and at response assessment [4,19].
| Category | Bone marrow criteria1 | Peripheral blood criteria1 | Serologic criteria2 | Other criteria |
|---|---|---|---|---|
| Stringent complete remission (sCR) | – Bone marrow plasma cells <5% and | – No plasma cells in peripheral blood by flow cytometry | – Negative serum and urine immunofixation | – Absence of extramedullary disease |
| – No malignant plasma cell by flow cytometry | – Normal serum free light chain (FLC) ratio | |||
| Complete remission (CR) | – Bone marrow plasma cells <5% | – No plasma cells in peripheral blood | – Negative serum and urine immunofixation3 | – Absence of extramedullary disease |
| Very good partial response (VGPR) | – Bone marrow plasma cells <5% | – No plasma cells in peripheral blood | – ≥90% reduction of serum M-protein, and | – Absence of extramedullary disease |
| – 24-h urinary M-protein <100 mg per 24 h4 | ||||
| Partial response (PR) | – Bone marrow plasma cells to 5–25% | – Peripheral plasma cell from 1% to 5% | – ≥50% reduction of serum M-protein and | – ≥50% reduction in the size of extramedullary disease |
| – Reduction in 24-h urinary M-protein by ≥90% and <200 mg per 24 h5 | ||||
| Stable disease (SD) | – Not meeting the criteria of either partial response or progressive disease | |||
| Progressive disease (PD) | – >25% increase in plasma cells in a bone marrow aspirate or absolute increase ≥10% | – Plasma cells >5% absolute increase in peripheral blood | – >25% increase in the level of the serum monoclonal paraprotein with an absolute increase ≥5 g/L | – Hypercalcemia |
| – Definite increase in lytic bone lesions | ||||
| – >25% increase in the 24 h urinary light chain excretion with and absolute increase ≥200 mg/24 h | – Definite increase in the size or number of extramedullary disease | |||
| Relapse from CR | – More than 10% increase in bone marrow plasma cells | – Reappearance of peripheral blood plasma cells at any level | – Reappearance of original M-protein in serum and/or urine immunofixation | – Any extramedullary disease |
It is recommended that at least 200 leukocytes on blood smears and 500 nucleated cells on marrow smears be counted.
It should be maintained for a minimum of 6 weeks. In case of discrepancy or undetectable serological parameter, the patient must be classified according to bone marrow criteria.
If the serum and urine M-protein are unmeasurable, a normal serum kappa/lambda FLC ratio is also required.
If the serum and urine M-protein are unmeasurable, a ≥90% decrease in the difference between involved and uninvolved FLC levels is required instead of the M-protein.
If the serum and urine M-protein are unmeasurable, a ≥50% decrease in the difference between involved and uninvolved FLC levels is required instead of the M-protein.
Response criteria for plasma cell leukemia. Adapted from: Fernández et al. [4]
3.5. Treatment Outcomes
With conventional chemotherapy, the prognosis of pPCL has been ominous, with reported OS ranging from 6.8 to 12.6 months [13,15,53–55]. The survival rate at 5 years from the time of diagnosis is <10% in all reported case series. The best treatment outcome was a median survival longer than 3 years reported in the pPCL patients who underwent stem cell transplantation [56]. In the era of novel agents and the use of ASCT following induction therapy appears to be associated with deeper response and an improved, though still poor, survival [15,17]. An epidemiological study of 445 patients with pPCL from a US registry showed significant improvement in OS, from 5 months reported between 1973 and 2005 to 12 months in the 2006–2009 period [53].
With respect to upfront ASCT, there are a few retrospective studies that support its superiority. The largest one, including 272 patients with pPCL, conducted by the EBMT [57], reported higher probability of achieving a complete remission after autologous transplantation in comparison to MM patients (41.2% vs. 28.2% at 100 days post-transplantation, respectively; p = 0.000). Conversion from a less than complete remission to CR improved the OS of PCL patients (HR = 0.59; 95% CI, 0.34–1.05); however, it still remained significantly worse than MM patients achieving CR in the same period (HR = 3.18; 95% CI, 2.55–3.96). The median OS in pPCL was inferior to that in MM (25.7 months, 95% CI, 19.5–31.9 months vs. 62.3 months, 95% CI, 60.4–64.3 months, respectively) (p = 0.000), probably as a consequence of the short response duration and increased relapse-related mortality observed in the first group. A study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) reported a progression-free survival (PFS) and OS at 3 years of 34% and 62%, respectively, in 97 patients with pPCL who underwent ASCT [56]. These results demonstrated for the first time survival beyond 3 years in selected individuals, supporting the IMWG recommendation for intensive treatment whenever there are no contraindications related to age and general clinical condition [4]. The data, although promising to some extent, are based only on retrospective analysis, without a direct group comparison.
As for novel antimyeloma agents, in an updated CIBMTR and Surveillance, Epidemiology, and End Results Program database combined analysis [58], it was noted that a higher proportion of newly diagnosed pPCL patients received modern induction regimens and were able to undergo auto- or allotransplantation in recent years (2008–2015). However, this did not translate into a significant additional posttransplant benefit. The outcomes remained dismal, with a 4-year PFS of 17% (13–23%), and OS of 28% (22–35%). Posttransplant relapse remains one of the biggest challenges.
In summary, taking into consideration the outcomes of pPCL patients treated frontline with novel agents [15,17,27,28,40–42,59–64], following Musto et al. [36] (Table 2), particularly bortezomib and lenalidomide induce an overall response rate (ORR) ranging from 57% to 87%, with up to 45% CR. The OS in young patients undergoing transplantation ranges from 16 to 61 months, while in elderly transplant-ineligible individuals it varies between 12 and 28 months.
| References | Patients [N] | ASCT [%] | Novel agents [%] | ORR with novel agents [%] | CR with novel agents [%] | Median PFS with novel agents [months] | Median OS with novel agents |
|---|---|---|---|---|---|---|---|
| Prospective trials | |||||||
| Musto et al. [40] | 23 | 39 | 100 | 74 | 13 | 14 (27 with ASCT vs. 2 w/o ASCT) | 28 (not reached at 34 months with ASCT vs. 12 w/o ASCT) |
| Royer et al. [49] | 39 | 100 | 100 | 69 | 10 | 15.1 (not reached at 28.7 months with double ASCT vs. 17.9 with tandem ASCT/AlloSCT) | 36.3 (not reached at 28.7 months with double ASCT vs. 36.3 with tandem ASCT/AlloSCT) |
| Retrospective and observational studies | |||||||
| Iriuchishima et al. [23] | 38 | NA | 61 | 67 | NA | NA | 34.2 (53.4 with maintenance vs. 34.2 w/o maintenance after ASCT) |
| Jung et al. [27] | 59 | 37 | 61 | 75 | 22 (after induction)/32% (after ASCT) | 12.9 (26.4 with ASCT vs. 9 w/o ASCT) | 18.5 (31.1 with ASCT vs. 12.3 w/o ASCT) |
| Katodritou et al. [30] | 50 | 32 | 80 | 82 | 18 | 12 (18 with btz + ASCT vs. 9 in others) | 18 (48 with btz + ASCT vs. 14 in others) |
| Jurczyszyn et al. [29] | 117 | 52 | 92 | 79 | 21 | NA | 23 (35 with ASCT vs. 13 w/o ASCT) |
| Ganzel et al. [14] | 37 | 51 | 85 | 57 | 22 | NA | 22.5 (all patients, excluding early deaths; 35.5 with ASCT) |
| Nakaya et al. [42] | 23 | 39 | 87 | 75 | 0 | NA | 34 (40 with ASCT, 55 with AlloSCT, 61 with tandem ASCT/AlloSCT vs. 28 w/o any transplant) |
| Mina et al. [37] | 38 | 74 | 100 | 87 | 45 | 20 (25 with ASCT vs. 6 w/o ASCT; 27 with ASCT and maintenance vs. 11 with ASCT w/o maintenance) | 33 (38 with ASCT and maintenance vs. 22 with ASCT w/o maintenance) |
| Gowda et al. [19] | 23 | 100 | 100 | 65 | 13 (after induction)/26 (after ASCT) | 5.5 (18.6 with maintenance vs. 3.8 w/o maintenance) | 18.1 (31.8 with maintenance vs. 16.1 w/o maintenance) |
AlloSCT, allogeneic stem cell transplantation; ASCT, autologous stem cell transplantation; btz, bortezomib; CR, complete response; NA, not available; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; VGPR, very good partial response.
Treatment outcomes in most recent studies on pPCL patients treated frontline with novel agents. Adapted from: Musto et al. [36]
The unfavorable prognosis in pPCL results from the very aggressive course of the disease, with several complications leading to a high percentage of early deaths within the first months from diagnosis, and from the lack of effective treatment options, which precludes the achievement of deep and prolonged responses. More clinical trials are needed in this high-risk patient group.
3.6. Prognostic Factors
There are a few clinical and laboratory factors considered as unfavorable for MM, including elevated LDH and β2-microglobulin, low serum albumin and high-risk cytogenetic changes. They also have a negative impact in pPCL, but occur less frequently than in MM, in which they present no prognostic significance [36].
In the phase II trial designed by the Intergroupe Francophone du Myélome assessing the outcomes of treatment consisting of bortezomib, doxorubicin, cyclophosphamide and dexamethasone followed by stem cell transplantation, none of the genetic features analyzed in the study were prognostic of survival. MYC protooncogene rearrangements assessed by single nucleotide polymorphism were associated with poorer outcomes [27].
Real-world data from the multicenter national study, conducted by the Greek Myeloma Study Group, so far one of the largest reported national series of 50 pPCL patients treated mostly with novel agents, showed that achievement of at least very VGPR and LDH < 300 U/L were significant predictors for OS [62].
In a multicenter retrospective study that analyzed clinical characteristics and outcomes in 117 patients with pPCL [17], Jurczyszyn et al. presented three independent predictors of worse survival: age ≥60 years, platelet count ≤100 × 109/L and peripheral blood plasma cell count ≥20 × 109/L. The combination of those parameters forms a PCL prognostic index (PCL-PI) that helps to identify patients with significantly different outcomes, as shown by OS at the levels of 46, 27 and 12 months for those with 0, 1 or 2–3 of these risk factors, respectively (p < 0.001) (Figure 4). The three variables included in the PCL-PI can be easily measured and are routinely determined during diagnostics. Therefore, in comparison to less accessible cytogenetics or immunophenotyping methods, the index may be a useful tool in identifying a group of patients who may particularly benefit from more aggressive treatment [17].
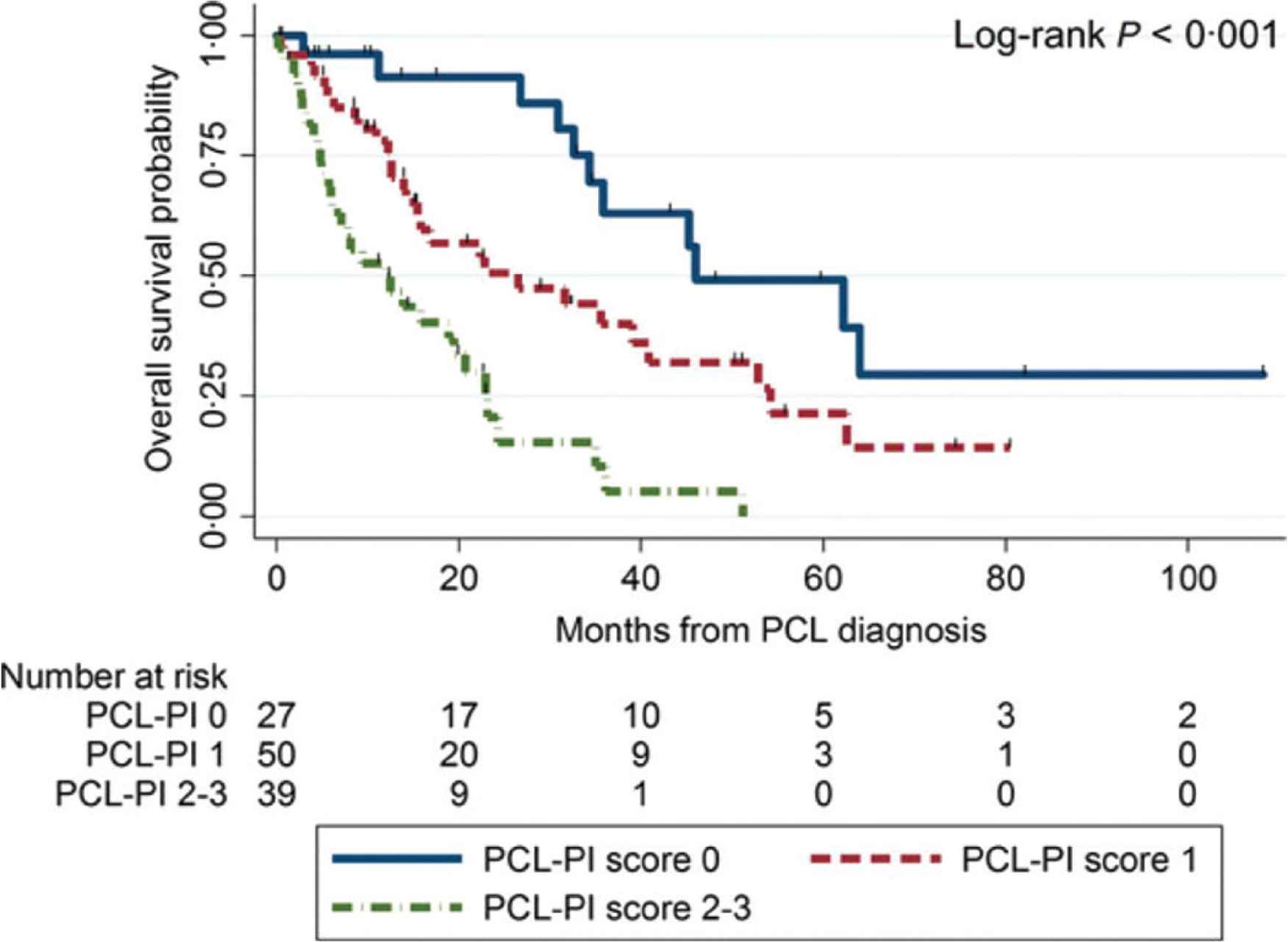
Overall survival estimates in 117 patients with primary plasma cell leukemia by pPCL-PI. Adapted from: Jurczyszyn et al. [17]. PCL-PI, plasma cell leukemia prognostic index.
4. SECONDARY PLASMA CELL LEUKEMIA – BRIEF PRESENTATION
Most of the data regarding the natural history of sPCL and its treatment outcomes has been derived from individual case reports and small case series [38,65–67]. sPCL is a rare entity, occurring approximately in 1% of all MM (up to 12% of those with high tumor burden), after a median time of about 31 months of the course of the disease [16]. It develops in patients approximately 10 years older than those with pPCL.
Among all symptoms, the presence of osteolytic lesions is higher than in pPCL [13]. Patients with sPCL evolving from MM usually present with normal or moderately increased serum LDH level. A significant elevation in serum LDH level was observed in those with high tumor burden [68], although without clear prognostic value. Notably, in the univariate analysis from a multicenter retrospective study, platelet count <100 × 109/L at the time of sPCL diagnosis was estimated to be the only independent predictor of a poorer OS (HR = 3.98, p = 0.0001). However, thrombocytopenia is a common finding in sPCL (almost 80% of patients present with platelet counts <100 × 109/L), which practically makes this factor clinically irrelevant [16].
Since sPCL is the final evolution of MM, all available antimyeloma therapies have usually been used, including ASCT [32]. The therapeutic approaches depend on the type of and response to previous MM therapy. If possible, fit individuals may undergo intensive chemotherapy with bortezomib-based regimens, which may slightly improve the adverse outcome for these patients. However, sPCL, as a terminal phase of MM, has an extremely poor prognosis with <50% response to treatment, and a median OS of 1 month [4]. Based on observations from a multicenter retrospective study in 101 sPCL patients (with median age of 62 years), the use of salvage multidrug PI-based regimens and ASCT may provide response and survival benefits [16].
5. EMERGENCIES AND SUPPORTIVE TREATMENT IN PCL
Similar to acute leukemias, the progression of PCL is very rapid. Thus, tumor lysis syndrome (TLS) associated with high tumor burden and elevated proliferative index may occur. Serum uric acid, calcium, phosphorous and creatinine levels must be monitored regularly. Standard supportive treatment in all PCL patients should include bisphosphonates, TLS monitoring and prevention, antiviral and antibiotic prophylaxis akin to MM recommendations. Thromboprophylaxis should be considered in patients treated with IMIDs because of their increased risk of venous thromboembolism.
6. CONCLUSION
Plasma cell leukemia is a highly aggressive neoplastic entity among monoclonal gammopathies. An arbitrary diagnostic approach, including absolute number of circulating PCs exceeding 2.0 × 109/L and/or >20% PCs in the total leucocyte count, may underestimate the real clinical significance of circulating PCs, thus, the current definition is under debate. Treatment should ensure a rapid cytoreduction leading to disease control in order to reduce the risk of early death. However, optimal therapy still remains an unmet clinical need. The recommendations concerning therapeutic approaches in PCL are supported by limited data and based mainly on expert opinion. Clinical trials and genomic studies may contribute to create more personalized schemas and improve the outcomes. So far, there seems to be more questions than answers in the context of PCL.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
All authors made substantial contributions to the present article. AS conceptualized this manuscript. All the authors were responsible for the literature search, analysis and interpretation, and wrote the paper. MS prepared microscope images and their captions. AJ and DHV revised the manuscript before submission.
FUNDING
No financial support was provided.
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - Anna Suska AU - David H. Vesole AU - Jorge J. Castillo AU - Shaji K. Kumar AU - Hari Parameswaran AU - Maria V. Mateos AU - Thierry Facon AU - Alessandro Gozzetti AU - Gabor Mikala AU - Marta Szostek AU - Joseph Mikhael AU - Roman Hajek AU - Evangelos Terpos AU - Artur Jurczyszyn PY - 2020 DA - 2020/07/19 TI - Plasma Cell Leukemia – Facts and Controversies: More Questions than Answers? JO - Clinical Hematology International SP - 133 EP - 142 VL - 2 IS - 4 SN - 2590-0048 UR - https://doi.org/10.2991/chi.k.200706.002 DO - 10.2991/chi.k.200706.002 ID - Suska2020 ER -