
What’s Important and New in Hemochromatosis?
- DOI
- 10.2991/chi.k.200726.001How to use a DOI?
- Keywords
- iron overload; hepcidin; ferroportin; non-transferrin bound iron; HFE; hemojuvelin; transferrin receptor 2
- Abstract
Major advances in the understanding of genetic iron overload have led to a clarification of the nosology and terminology of the related diseases. The term hemochromatosis should be reserved to the entities where iron overload is related to hepcidin deficiency or hepcidin resistance. The diagnosis of hemochromatosis is non-invasive, based on clinical examination, blood investigations and, whenever possible, magnetic resonance imaging. Phlebotomies remain the mainstay of the treatment, but new therapeutic approaches should, in the future, constitute a valuable advance, hopefully both as an adjunct to bleeding in the induction phase and as its replacement in the maintenance phase. The goal of the present review is to update the terminology of hemochromatosis in light of major pathophysiological advances, and the main features of its diagnostic and therapeutic approaches.
- Copyright
- © 2020 International Academy for Clinical Hematology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
The goal of the present review is to update the terminology of hemochromatosis in light of major pathophysiological advances, and the main features of its diagnostic and therapeutic approaches. This article is the basis of the webinar organized by the International Academy for Clinical Hematology on June 10, 2020 (http://clinical-hematology.org/video-library/).
2. UPDATE ON HEMOCHROMATOSIS DEFINITION
Based on the conclusions of the international workshop devoted to this topic during the BioIron Congress held in Heidelberg in 2019 (manuscript in preparation), hemochromatosis can be defined as a chronic iron overload where the development of iron excess is due to hepcidin deficiency or hepcidin-resistance.
2.1. Focused Reminders on Iron Metabolism
Hepcidin is the iron hormone that regulates systemic iron metabolism [1,2] (Figure 1). It is a small peptide, mainly produced by hepatocytes, whose function is to decrease plasma iron concentration. This decrease is mainly the result of the increased intracellular degradation of ferroportin, which is today the only known protein involved in the iron export from cells into the plasma. The main impact of hepcidin on ferroportin takes place at two main sites, the duodenocyte and the splenic macrophage. As a result of this hepcidin action, the plasma iron concentration decreases by impairing both duodenal iron absorption and the release of splenic iron which comes from the degradation of senescent erythrocytes. Therefore, diminished hepcidin hepatic synthesis leads to hypohepcidinemia that, in turn, produces hypersideremia via digestive hyperabsorption of iron and increased iron recycling from the spleen. Once into the plasma, the metal is taken up by transferrin, whose capacity of carrying iron (total iron-binding capacity) is in excess of the physiological iron concentration, corresponding to the clinical notion of partial transferrin saturation (TfSat) which is normally <45%.
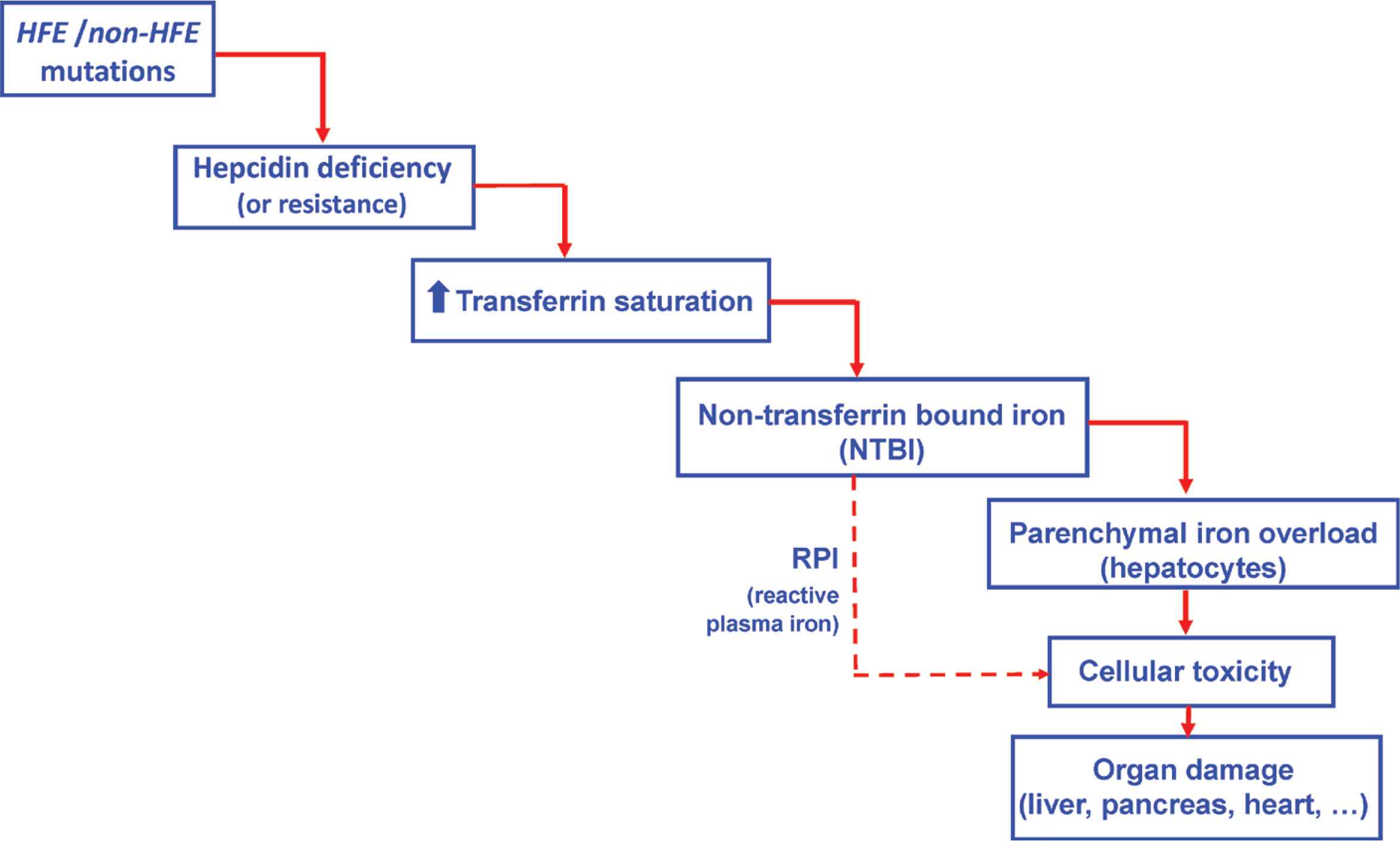
Hemochromatosis: mechanistic cascade from mutation to organ damage.
A further aspect of the hepcidin–ferroportin interaction needs to be considered. Ferroportin is not only an iron export protein but also acts as the membrane receptor for plasma hepcidin. This is why in some genetic situations where ferroportin’s receptor function is impaired, plasma hepcidin, despite being in normal concentrations, may be unable to bind the ferroportin receptor. This corresponds to hepcidin resistance, whose final effect is similar to that of decreased plasma hepcidin concentrations, namely an increased activity of the iron export property of ferroportin (the export function is maintained despite the mutations impacting the ferroportin receptor property).
Whatever the mechanism whereby cellular lack of hepcidin leads to increased iron concentration in the plasma, the fate of this excess plasma iron is to be uptaken by plasma transferrin. The result is an increased saturation of transferrin. When the TfSat exceeds 45%, a new plasma iron form may appear that is no more linked to transferrin and is called non-transferrin bound iron (NTBI) [3]. In contrast to transferrin-bound iron whose main target is the bone marrow (where it is used to produce new red blood cells), NTBI is very avidly taken up by parenchymal cells and, initially, by hepatocytes. Furthermore, whenever the TfSat is over 75%, a peculiar plasma NTBI species may be present, named reactive plasma iron (RPI) or labile plasma iron [4]. RPI is defined by its propensity to produce reactive oxygen species known to exert deleterious effects on cell plasma membranes as well as on membranes of intracellular organelles. Therefore, RPI is considered as the potentially toxic form of plasma iron, responsible for cell death followed by damage to organs such as the liver, pancreas or heart.
2.2. Definition of Hemochromatosis
Hemochromatosis consists of a series of genetic iron overload diseases in which iron excess is due to hepcidin deficiency or to a hepcidin refractory state (Figure 2).
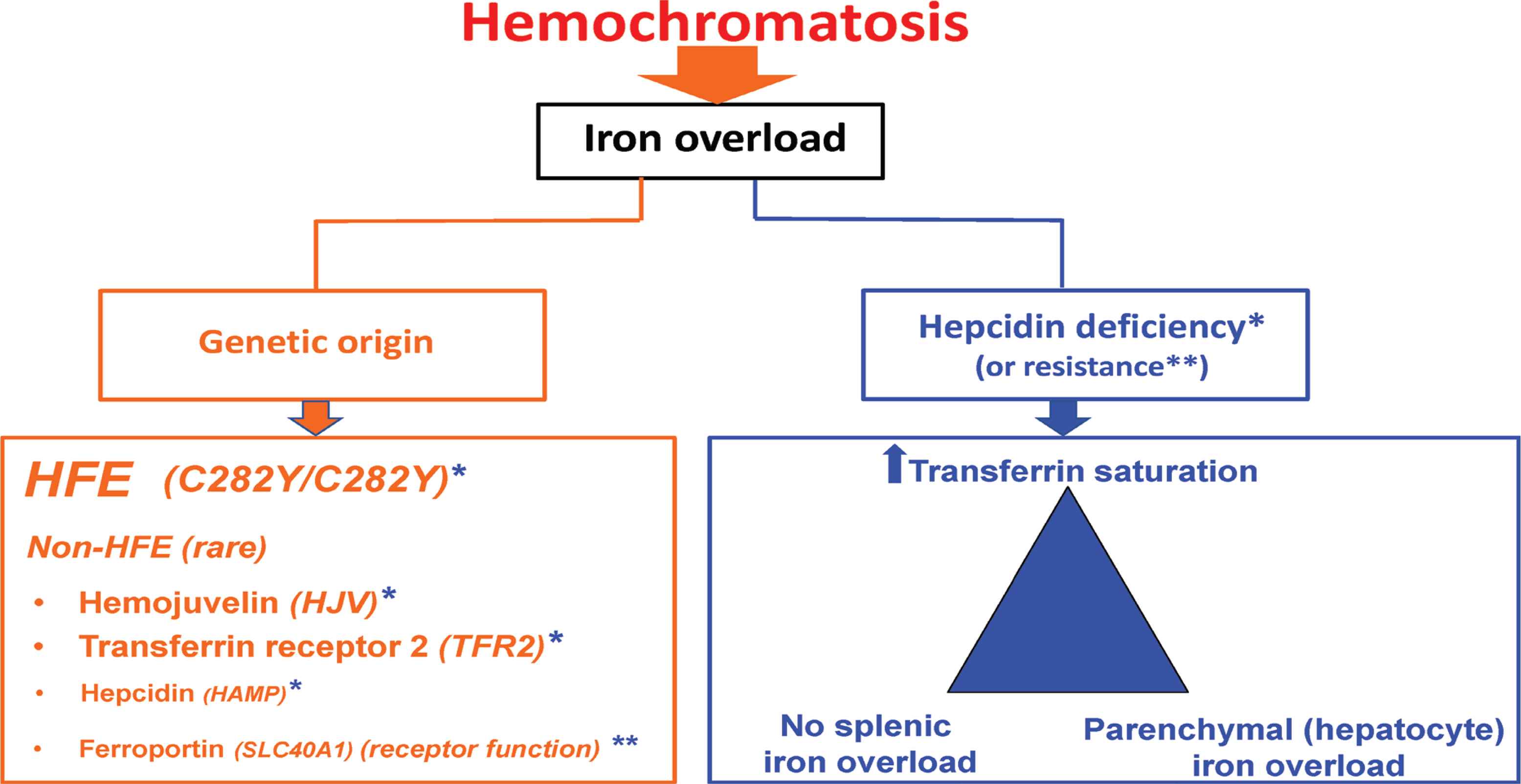
Hemochromatosis: updated definition. *refers to hepcidin deficiency; **refers to hepcidin resistance.
In hemochromatosis related to hepcidin deficiency both HFE and non HFE mutations can be involved [5]. HFE-related hemochromatosis is, by far, the most frequent form of hemochromatosis. It is mainly due to homozygosity for the C282Y mutation that is present in more than six subjects out of 1000 in the Caucasian population [6]. Exceptional forms of compound heterozygosity (C282Y plus another rare HFE mutation) may also be involved. Non HFE-related hemochromatosis is caused by rare mutations of genes that code for proteins involved in the hepcidin synthetic pathway such as the hemojuvelin (HJV), hepcidin (HAMP), and transferrin receptor 2 (TFR2) genes.
Hemochromatosis related to hepcidin resistance is a rare form of hemochromatosis due to mutations of the ferroportin gene (SLC40A1) that impair the hepcidin-receptor function of ferroportin [7].
The hepcidin deficiency (or resistance) syndrome is shared by the different types of hemochromatosis, and corresponds to the following triad: increased plasma transferrin saturation, parenchymal (mainly hepatocyte) iron overload, and no spleen iron overload.
2.3. What is not Hemochromatosis
Two categories of non-hemochromatosis iron overload disorders can be identified.
2.3.1. Acquired iron overload
It may be due to multiple transfusions (ß-thalassemia major [8], myelodysplactic syndromes [9]), to excessive parenteral iron supplementation (chronic digestive bleeding, chronic renal failure [10]) or to ineffective erythropoiesis [11]. In the latter setting, it should be pointed out that, as for hemochromatosis, the mechanism for the development of iron excess is hepcidin deficiency, since dyserythropoiesis increases the bone marrow production of the hormone erythroferrone, which acts by decreasing hepcidin synthesis [12]; this explains why iron overload, resembling that of hemochromatosis, can occur in these hematological situations in the absence of any transfusion.
2.3.2. Non-hemochromatosis genetic iron overload
In this situation, iron overload, although of genetic origin, is due to mechanisms that do not involve hepcidin deficiency or resistance. This includes: (i) the ferroportin disease [13] where causal mutations that hamper the cellular export capacity of iron, and (ii) hereditary aceruloplasminemia [14] whose mechanism of iron excess is likely more complex than the initially suggested indirect impairment (via the absence of ferroxidase activity) of the ferroportin iron export property [15].
In short, this new terminology renders obsolete many formulations that are still overused in the field of iron overload, such as primary hemochromatosis, genetic hemochromatosis, hereditary hemochromatosis, secondary hemochromatosis, or hemosiderosis.
3. UPDATE ON HEMOCHROMATOSIS DIAGNOSIS
3.1. HFE-related Hemochromatosis
The main notion is that the diagnosis is now based on a totally non-invasive approach, meaning that a liver biopsy is no longer needed to establish it [5].
The diagnosis is based on a triad consisting of clinical, biological, and imaging data.
3.1.1. Clinical data
It is characterized, in a Caucasian individual, by a mute, delayed, and often confusing expression. It should first be recalled that, the disease can be totally silent for 30–50 years, corresponding to the very slow process of body iron deposition. This long “black box” phase explains why the diagnosis cannot be made in the absence of systematic control of plasma iron parameters (TfSat and ferritin). When clinical signs emerge, they correspond nowadays to a changing picture as compared to the classical initial description of the “bronzed cirrhosis with diabetes”. Presenting symptoms, alone or combined, consist of chronic unexplained fatigue, often misinterpreted as being of psychological origin, and arthropathy, misinterpreted as arthrosis or rheumatoid arthritis. Two localizations should suggest hemochromatosis arthropathy: the hands (with the symptom of “painful handshake”) and the ankles. Often, early osteoporosis is also present. Later on in the course of disease, syndromes may develop that not only impair the quality of life but may be life-threatening: liver cirrhosis (with the risk not of hepatic failure but of hepatocellular carcinoma), insulin-dependent diabetes, and, more rarely, cardiac signs such as arrythmias and, eventually, heart failure.
3.1.2. Biological data
3.1.2.1. Plasma transferrin saturation
As previously mentioned, plasma TfSat is the earliest biochemical abnormality in hemochromatosis, so that normal TfSat rules out the diagnosis. It is usually >60% in men and >50% in women, and frequently close to 100%. It should be checked twice, given that it is a rather fluctuating parameter. It is important to explain to the patient that a TfSat value of 80% does not mean that the body is saturated at 80% but that only the blood iron shuttle, transferrin, is full at 80% of its transport capacity. It should however be kept in mind that TfSat values permanently over 75% may have some prognostic meaning (since it may be associated with the presence of RPI in the plasma).
3.1.2.2. Plasma ferritin
Its value is usually >300 ng/mL in men and >200 ng/mL in women. It reflects the amount of increased body iron load, provided the three other main mechanisms of hyperferritinemia, which are not related to iron overload, have been excluded: metabolic syndrome, inflammation, and chronic alcoholism. Iron excess is considered as moderate up to 500, important between 500 and 1000, and major above 1000 ng/mL.
3.1.2.3. Genetic test
The expected result for establishing the diagnosis of HFE-hemochromatosis is homozygosity for the C282Y mutation (C282Y/C282Y), one mutation coming the father, the other from the mother (who are typically both heterozygote and, therefore, not affected, hemochromatosis being a recessive disease). The presence of H63D heterozygosity, H63D homozygosity, or the S65C mutation are not relevant for the diagnosis of hemochromatosis. The interpretation of compound heterozygosity involving the C282Y mutation is more complex. Rare genetic profiles of compound heterozygosity can be responsible for the development of the disease, but this is not the case for the frequent C282Y/H63D compound heterozygosity. It is widely accepted that, if this genetic profile can cause a significant increase in plasma TfSat (usually <70%), it is not by itself responsible for clinically significant body iron overload. In fact, this form of compound heterozygosity may have more of a “boosting” effect for increasing ferritinemia, when the main mechanism involved in this ferritin increase corresponds to the situations previously mentioned (especially the metabolic syndrome) [16].
3.1.3. Imaging data
The input of magnetic resonance imaging (Iron-MRI) has revolutionized the diagnosis of iron overload [17]. In the case of hemochromatosis, it presents a triple interest: (i) to visualize hepatic iron excess (iron decreasing specifically the MRI signal); (ii) to quantify this excess (the darker the liver, the higher the hepatic iron concentration); (iii) to provide an important clue for the mechanism of iron overload by assessing the respective hepatic and spleen iron load. Thus, in hemochromatosis, the typical picture will be the contrast between a “dark” liver (“black” if iron excess is massive) and a “white” spleen (Figure 3).
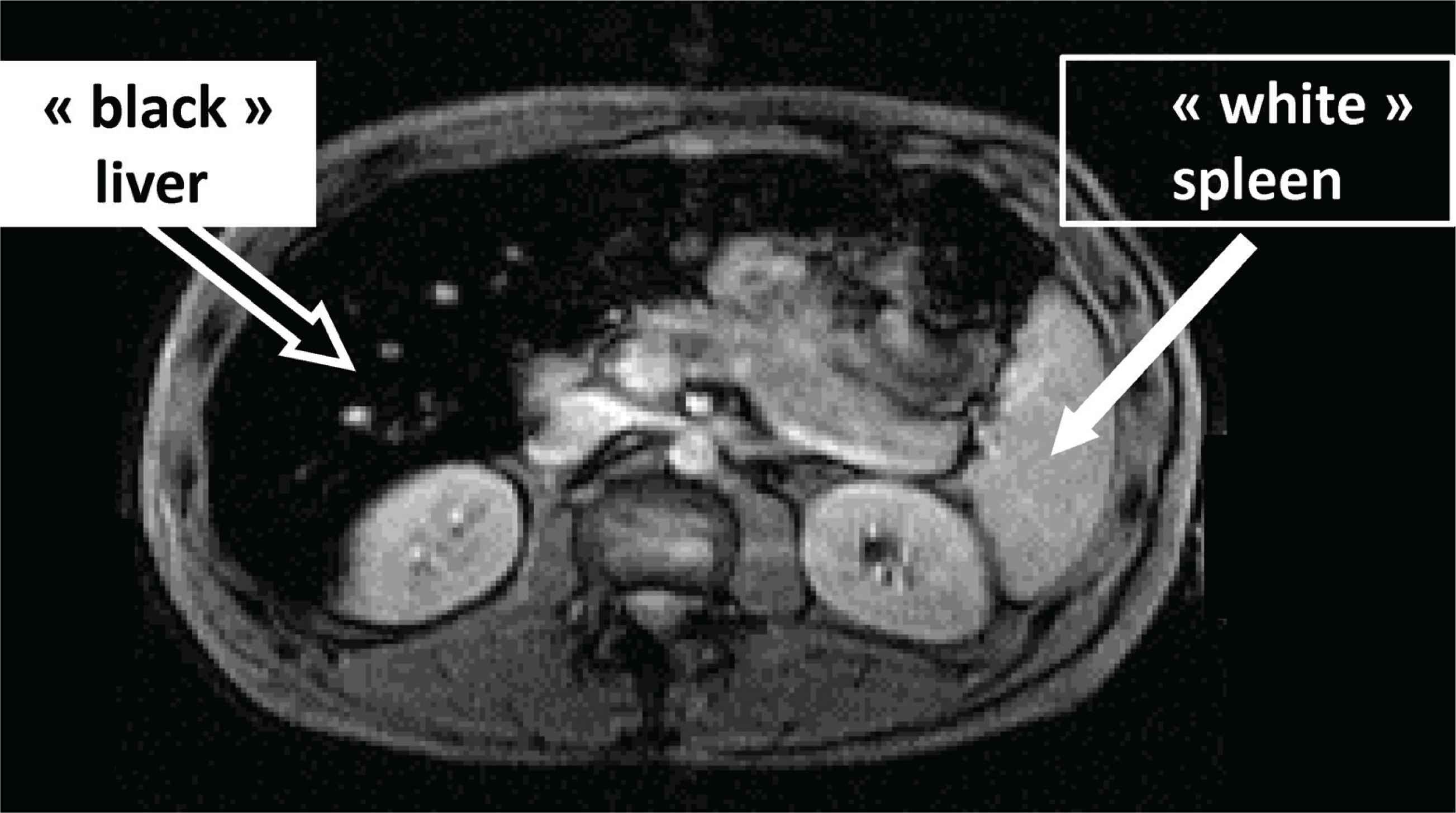
Typical magnetic resonance imaging in hemochromatosis (massive hepatic iron overload contrasting with ‘non-overloaded spleen’).
3.2. Non HFE-related Hemochromatosis
They are due to mutations of the following genes: HJV, HAMP, TFR2 or SLC40A1 (restricted here to mutations targeting the receptor function of ferroportin). Since iron overload is, like in HFE hemochromatosis, the consequence of cellular hepcidin deprivation, non-HFE hemochromatosis will present close similarities to HFE-hemochromatosis corresponding to the hepcidin deficiency triad: increased plasma TfSat, parenchymal (hepatocyte) iron overload, and no splenic iron overload. But there are some differences with HFE-hemochromatosis: these diseases are much rarer; they are multi-ethnic (i.e. not limited to Caucasians); they occur in younger subjects (under 30 years of age); they are more severe, especially in their hepatic and cardiac complications; they require to be genetically diagnosed by expert laboratories and practically managed by expert reference centers.
3.3. Non-hemochromatosis Genetic Iron Overload
3.3.1. Ferroportin disease
Being only form of genetic iron overload with a dominant pattern of transmission, the ferroportin disease is the most frequent of these rare non hemochromatosis genetic iron overload disorders [18]. The causal mutations impair the export function of ferroportin (especially at the macrophage level), leading to prevailing splenic iron excess with relatively moderate hepatic iron overload. Thus, organ iron excess is due to decreased egress of cellular iron and not (as for hemochromatosis) to increased entry of NTBI into cells. This major pathophysiological difference explains the point-by-point differences with the “hepcidin triad”: no increased plasma transferrin saturation, moderate hepatic iron excess, major splenic iron overload (therefore a “black” spleen and a “grey” liver at iron-MRI). Moreover, in the absence of NTBI, the ferroportin disease is globally less damaging than hemochromatosis.
3.3.2. Hereditary aceruloplasminemia
The presenting picture may be hematological with a microcytic, hyposideremic anemia, at first suggesting iron deficiency, but with an unexpected (often sharp) hyperferritinemia. The presence of neurological signs (due to brain iron overload) is a valuable diagnostic clue; it is the only genetic systemic iron overload disease with neurological complications. Once evoked, the diagnosis is easily made by checking the concentration of plasma ceruloplasmin that is not detectable. Genetic testing should only be performed after aceruloplasminemia has been biochemically confirmed. The pathophysiological explanation of iron overload remains to be clarified, since iron overload distribution mimics that of hemochromatosis (“black” liver and “white spleen”), whereas the classical proposed mechanism should lead more to a ferroportin disease-like distribution.
These two differential diagnoses of hemochromatosis illustrate the notion that body iron overload can be present despite normal or low TfSat levels.
4. UPDATE ON HEMOCHROMATOSIS TREATMENT
On this section, we will only consider the treatment of iron overload.
4.1. Phlebotomies
Bloodletting remains the mainstay of treatment because of its good overall tolerance, simplicity, low cost and effectiveness (except on joint symptoms). During the induction phase, usually consisting of weekly venesections, the appropriate marker of efficiency is ferritinemia, whose concentration is well correlated with body iron stores, the goal being to reach approximately a level of 50 ng/mL. In contrast, plasma TfSat is not a reliable marker, since it remains quite elevated during most of this phase, and drops (rather suddenly) only when ferritinemia is close to 50 ng/mL. During maintenance therapy the aim is to avoid the progressive reconstitution of iron overload. Three to six yearly phlebotomies may be needed, depending on each patient. The ideal biochemical profile follows the “rule of the fifty”, alluding to ferritinemia and TfSat around 50 ng/mL and 50%, respectively. However, this goal is sometimes difficult to achieve, with a subset of patients presenting high TfSat levels (over 75%) despite “correct” ferritinemia. When prolonged, this discrepancy may be clinically significant, with a higher proportion of fatigue and arthropathy [19]. Therefore, despite the fact that maintaining plasma ferritin concentration close to 50 ng/mL remains the key objective (it makes possible to assert the absence of any visceral iron overload), checking TfSat, for instance twice a year, may be a wise recommendation.
4.2. Other Treatments
4.2.1. Oral chelation
The oral chelator deferasirox has been reported to be efficient in hemochromatosis [20], through its capacity to remove iron both at the organ level (especially the liver) and at the plasma level (NTBI). However, apart from possible digestive, renal or hepatic side effects (which, as a rule, are moderate), its indications arising from contraindications to phlebotomies (difficulties in venous access, psychological intolerance) remain exceptional. Moreover, it is an off-label drug.
4.2.2. Innovative pathophysiological approaches
Despite its major interests, bloodletting is only a symptomatic treatment that essentially removes established iron overload at diagnosis and prevents its reconstitution after the induction phase. More rational approaches, stemming from advances in pathophysiological understanding, are therefore warranted. So far, two main options are under investigation. One consists in hepcidin supplementation, especially by subcutaneous injections of synthetic forms of hepcidin. The other is to counteract ferroportin activity, especially by using an oral ferroportin antagonist at the duodenal level, in order to decrease intestinal iron hyperabsorption. Proof of concept for both approaches has been provided in animals and healthy volunteers [21,22]. However, parenteral hepcidin supplementation was discontinued due to mixed results at the interim report of the only Phase II study reported to date in hemochromatosis subjects; ferroportin antagonists have not yet been tested in hemochromatosis.
5. CONCLUSION
In conclusion, major advances in the understanding of genetic iron overload have led to a clarification of the nosology and terminology of the related diseases. The term hemochromatosis should be reserved to the entities where iron overload is related to hepcidin deficiency or hepcidin resistance. The diagnosis of hemochromatosis is non-invasive, based on clinical examination, blood investigations and, whenever possible, MRI imaging. Phlebotomies remain the mainstay of the treatment, but new therapeutic approaches should, in the future, constitute a valuable advance, hopefully both as an adjunct to bleeding in the induction phase and as its replacement in the maintenance phase.
CONFLICTS OF INTEREST
PB has been a consultant for Novartis and La Jolla Pharmaceutical Company, and is presently consultant for Ionis pharmaceutical and Protagonist Therapeutics; EB has received funding from Novartis.
AUTHORS’ CONTRIBUTION
Both authors have contributed to the design and writing of the manuscript, and have approved its final version.
ACKNOWLEDGMENTS
The authors wish to thank the patient associations Association Hémochromatose Ouest (AHO) and Fédération Française des Associations de Malades de l’Hémochromatose (FFAMH) for their valuable contribution in increasing awareness of hemochromatosis and in improving the diagnostic and therapeutic management of the disease.
REFERENCES
Cite this article
TY - JOUR AU - Pierre Brissot AU - Eolia Brissot PY - 2020 DA - 2020/08/05 TI - What’s Important and New in Hemochromatosis? JO - Clinical Hematology International SP - 143 EP - 148 VL - 2 IS - 4 SN - 2590-0048 UR - https://doi.org/10.2991/chi.k.200726.001 DO - 10.2991/chi.k.200726.001 ID - Brissot2020 ER -