
Turner syndrome and guidelines for management of thoracic aortic disease: Appropriateness and utility
- DOI
- 10.1016/j.artres.2016.07.001How to use a DOI?
- Keywords
- Turner syndrome; Aortic aneurysm; Practice guidelines; Evidence-based practice
- Abstract
Introduction: Current guidelines for management of thoracic aortic disease base clinical decision-making primarily on the aortic size. The risk of serious complications in genetically-triggered aortic disease is influenced by many other factors, currently not taken into account by the guidelines.
Methods: An extensive review of the medical literature using PubMed and Medline.
Results: Current guidelines do not make a distinction between atherosclerotic and nonatherosclerotic aneurysmal disease. Current guidelines for genetically triggered thoracic aortic disease are based on data from Marfan syndrome population. There remain significant differences between Turner syndrome and Marfan syndrome (in patient gender, fertility, stature, female sex hormone and growth hormone therapy as well as presence of coarctation) may have influence on the natural history of the disease and risk assessment.
Conclusions: Turner syndrome patients have a distinct set of factors influencing the development and natural history of their aortic disease, as opposed to Marfan syndrome patients. Therefore, a different set of clinical, genetic and biomarker parameters need to be devised for this condition. Recommendations from the current guidelines need further updates to better address indications for surveillance and therapeutic interventions in patients with Turner syndrome.
- Copyright
- © 2016 Association for Research into Arterial Structure and Physiology. Published by Elsevier B.V. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
“All generalizations are false, including this one”
Background
The current clinical practice guidelines1,2 for management of thoracic aortic disease state that it is ‘reasonable’ to prophylactically replace the ascending aorta in asymptomatic patients with Turner syndrome at a diameter of 4–5 cm, since such patients are perceived to be a higher risk for aortic dissection or rupture. However, the 2014 revised European guidelines3 use the aortic diameter indexed by the body surface area as a threshold for intervention in these patients. This high level (Class I) recommendation is based on low-level supporting evidence (Level of Evidence C or “Expert Opinion”) The guidelines also alternatively include Turner syndrome as one of the so-called “connective tissue” disorders, or the “genetically mediated disorders”; similar to Marfan syndrome. We seek to examine the appropriateness4 of such recommendations in light of the existing body of scientific evidence.
Search methodology
An extensive literature search of the Medline, PubMed, National Institute of Medicine and the Royal Society of Medicine, UK databases was performed. Search items included: “Turner Syndrome”, “SHOX”, “Aortic Dissection” and combinations thereof.
Results
Incidence
Although the true epidemiology of Turner syndrome remains largely unknown, it is regarded as a rare disease. Its incidence has been reported with a high degree of variability (estimates ranging from 25 to 210 per 100,000 live-birth females), but it is generally thought to be around 1:2000.5–10 In fact, virtually all the available data is from either case reports or small-size series (only 3 cases in one report). Further complicating the issue11–13 are the following factors:
- -
Ascertainment bias, where the more severe cases are being reported based on post-natal diagnosis.
- -
The lack of systematic, comprehensive population-based epidemiologic studies
- -
Selection bias: Depending on the reporting specialty and researcher interest. Many cases are not recognized or misdiagnosed
- -
The late diagnosis in approximately 64% of cases, with recent study reports the average age of diagnosis to be 13–19 years.
- -
The high risk of in-utero death (usually during the first trimester) or termination of pregnancy (as high as 66% of cases)
- -
The high degree of phenotype variability, which may contribute to less severe cases being undiagnosed and/or under-reported.
Etiology and genetics
The genetics of Turner syndrome are still incompletely understood. The characteristic short stature in Turner syndrome has been linked to either an absence or haploinsufficiency of the ‘SHOX’ (Short Stature Homeobox containing gene) located on the short arm of the X sex chromosome (Xp), whose function remains unclear.14,15
The classic karyotype is 45, XO; seen in approximately 75% of cases and twice as likely to produce the phenotype with the more severe clinical findings. However, there is a high degree of variability of clinical presentation, due to mosaicism, in about 26% of cases. SHOX is primarily expressed in the developing distal limbs (in the osteogenic cells) and the 1st and 2nd pharyngeal arches.
SHOX deficiency mainly affects the developing limbs, leading to short stature, as in the classic picture of the Leri-Weill dyschondrosteosis. Absence of the X chromosome can have drastic effects on the developing embryo: The high incidence of intrauterine death16–18 has been attributed to fatal lymphatic dysplasia, with cystic nuchal hygromas developing at 10–12 weeks. In fact, the increased nuchal thickness on ultrasound examination in utero has been established as one of the key diagnostic findings in this syndrome. Surviving embryos have a higher incidence of left-sided primary heart tube developmental defects such as hypoplastic left heart syndrome, hypoplastic aorta or coarctation of the aorta. The lymphatic dysplasia associated with Turner syndrome has led to the postulation of a “lymphogenic gene” on the Xp arm that also escapes X-inactivation, which is responsible for the characteristic webbed neck and lymphedema seen in this condition.19–21 Our review of the current literature has failed to establish any link or association between SHOX and TGF-β ligand characteristic for Marfan syndrome and a number of other familial thoracic aortic diseases.22,23
Clinical picture
As the name implies, Turner syndrome describes a constellation of developmental failure exclusively in females, primarily affecting the skeletal and gonadal functions.
Skeletal features
Proportionate short stature is the hallmark of the condition, seen in over 90% of cases. Affected females are typically under 150 cm in height. Their length/height ratio is usually −1.7 Standard Deviations from the normal. This renders the use of absolute aortic diameters inappropriate in these patients. It is important to include leg measurements in the height analysis of these patients, given the proposed effects of SHOX on the long bones in the extremities. Importantly, here is no defect in the bone and cartilage metabolism or bone density itself,24–29 strongly arguing against any defect of the Extracellular Matrix structural proteins such as collagen.
Short stature in Turner syndrome is not due to deficiency of growth hormone. There is some evidence suggesting a mutation of the Insulin-like Growth Factor receptors (IGF-1) resulting in growth hormone-resistant growth retardation. This growth retardation actually starts in utero, and is first apparent by the first year of age.30–32 In fact, some current guidelines recommend growth hormone replacement therapy as early as 2 months of age.
Other skeletal manifestations include imbalanced upper extremity growth with cubitus valgus deformity (∼50% of cases), high-arched palate in 80%; and a characteristic broad or “shield” chest. However, scoliosis, kyphosis and other axial deformities classically seen in Marfan syndrome are absent.
Cardiovascular features
The many cardiovascular defects in Turner syndrome (Fig. 1 ) can be classified into the following main categories:
- -
Anatomic abnormalities of the developing thoracic aorta, specifically the development and differentiation of the aortic arch and its derivatives: This typically involves the third and fourth ‘aortic fields’. Aneurysmal dilatation of the descending aorta is very rare, and so are global aortic defects like diffuse dilatation or the mega-aorta à la Marfan syndrome. This is despite dilatation of some other major vessels such as the brachial or the carotid arteries, which share a common origin from the pharyngeal arches.
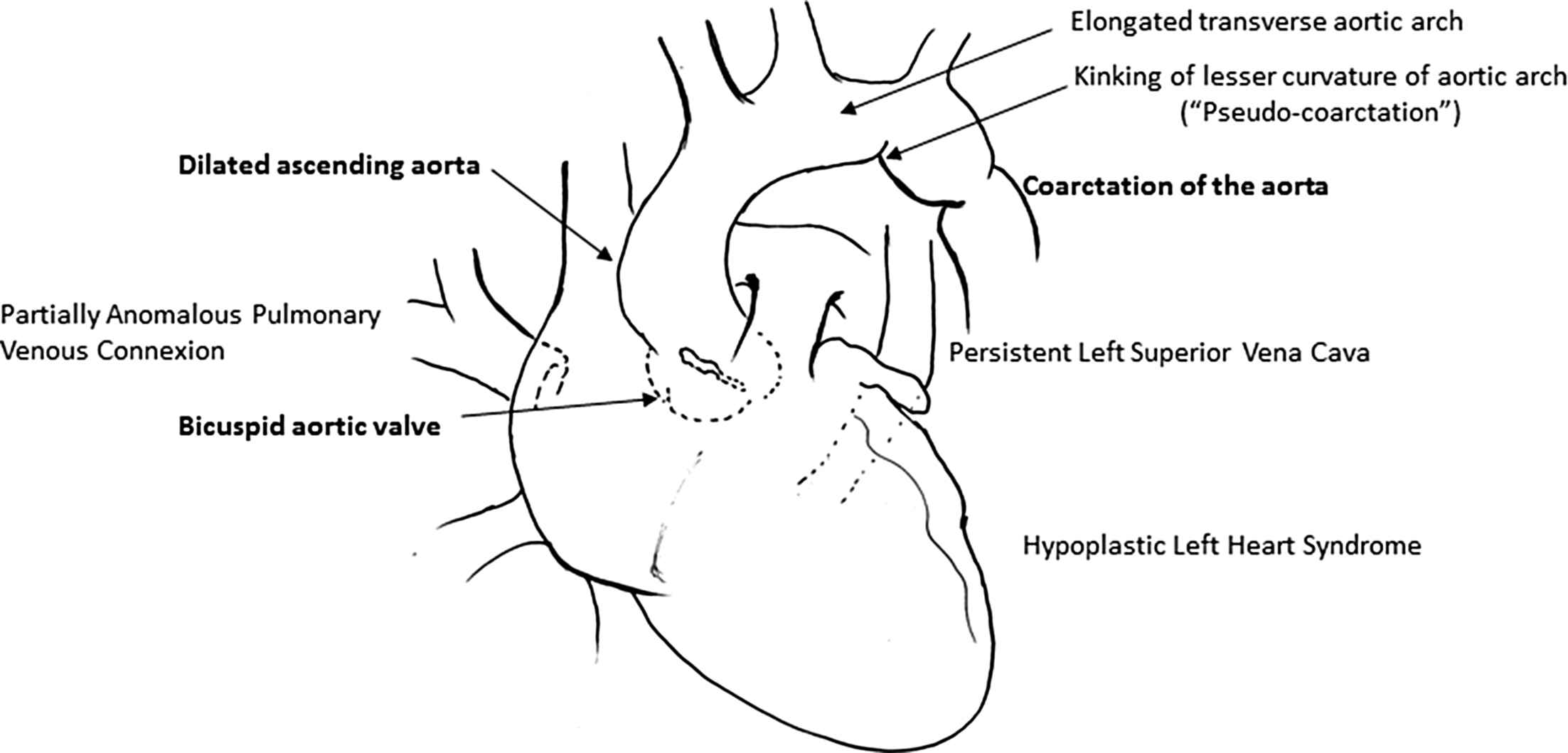
Diagrammatic representation of the common cardiovascular findings in Turner syndrome.
Subsequently, characteristic findings (Table 1 ) are primarily left-sided and involve an elongated transverse arch between the left common carotid and left subclavian artery (30% of cases), coarctation of the aorta in ∼13%, “pseudo-coarctation” or ‘kink’ at the aortic isthmus is seen in about 50%. A bicuspid aortic valve occurs in 30% and—curiously—hypoplastic left heart syndrome with aortic hypoplasia in 10% of cases.33–42 Currently, there are no longitudinal studies examining the natural history of aortic arch abnormalities.
| Systemic | Early-onset hypertension | 25% |
| Nocturnal non-dipping of blood pressure | ||
| Abnormal sympathetic nervous system function | ||
| Lymphedema | 60% | |
| Proximal ascending aorta | Bicuspid aortic valve | 27–30% |
| Dilated ascending aorta | 19–30% | |
| Aortic arch abnormalities (30–50% overall) |
Coarctation of the aorta | 8–12% |
| Elongated transverse arch | ∼30% | |
| Elongated zone 2–3 (distance between left common carotid and left subclavian artery) | ||
| Flattening of the transverse arch | ||
| Kinking along the lesser curve (pseudo-coarctation) | ∼50% | |
| Aberrant right subclavian artery | 9–16% | |
| Aortic and vascular wall properties | Increased Young’s elastic modulus | |
| Decreased wall elasticity (compliance) | ||
| Increased wall stiffness | ||
| Increased Pulse Wave Velocity | ||
| Increased Augmentation Index | ||
| Decreased carotid intima-media thickness | ||
| Congenital cardiovascular lesions | Partially anomalous pulmonary venous connection | 50% |
| Hypoplastic left heart syndrome | 10% | |
| Persistent left superior vena cava | 13% |
A list of the estimated incidence of common cardiovascular findings in Turner syndrome.
Pulmonary venous anomalies are also seen, with persistent left superior vena cava in 13% and partially anomalous pulmonary venous connexion in as many as 50%. Whether these lesions are due to a disturbance of left-right differentiation during development and differentiation of different aortic arch and conotruncal segments remains to be seen.42–46
The classic pathognomonic dilatation of the aortic annulus and aortic root (“annulo-aortic ectasia”) seen in almost every Marfan patient is characteristically absent or very rare. Dilatation of the mid-ascending aorta seems to correspond with the presence of a bicuspid aortic valve morphology.
- -
Other cardiovascular risk factors typically seen are out of proportion to the patient’s age. Turner syndrome females have a much higher incidence (7-fold increase) of premature coronary artery disease. They are typically hypertensive (25% of girls and 50% of adults), with a proatherogenic dyslipidemic blood lipid profile.46 There is a loss of the normal night-time decrease of the blood pressure; the so-called “dipping”. These changes are also associated with an abnormal sympathetic nervous system tone.48–50
Aortic and vascular wall mechanics exhibit changes similar to those seen in atherosclerotic disease. There is a decrease in aortic wall elasticity or compliance, leading to more ‘rigid’ and less distensible vessels. Pulse Wave Velocity and the Augmentation Index are increased, reflecting increased wall stress in the longitudinal axis. The Young’s Elastic Modulus is increased, reflecting the increased wall stress in the circumferential direction.45,51
Regarding aortic wall structure, current evidence demonstrates an increased intima-media thickness without endothelial dysfunction. An increased activity of ICAM adhesion molecule is suggested. There are no reports of specific molecular defects of collagen, elastin, fibrillin, fibrin or vascular smooth muscle cells or the relevant genetic and transcriptional pathways identified for genetically triggered aortic diseases. Flow-mediated dilatation, indicative of smooth muscle and tunica media function, is equal between Turner syndrome and normal individuals. In 2009, Dr Bondy emphasized “the lack of any actual evidence for any similar pathophysiology between Marfan and Turner syndromes”.
Left ventricular hypertrophy and a prolonged QT and QTc intervals have also been observed, possibly indicating an increase in myocardial fibrosis precipitating diastolic dysfunction.
These changes are almost the opposite of Marfan syndrome; the prototypical “connective tissue disease”.
Aortic dilatation as a precursor to aortic dissection has been postulated as a “primary aortic wall disease” without enough supporting evidence even to the site or degree of dilatation. The aortic growth rate in Turner syndrome (0.1–0.4 mm/year) is increased as opposed to the normal rate of 0.07 mm/year. However, there are currently no sufficient data evaluating the aortic growth in young patients with Turner syndrome.
In addition, aortic size does not correlate with the presence of a bicuspid aortic valve, and a limited study by Lanzarini52 suggests that the rate of progression is minimal at worst. In her state-of-the-art paper,53 Dr Bondy states that the raw value of ascending aortic diameter in Turner syndrome females was the same as control subjects, with the aortic size index of 1.8 in Turner syndrome patients as opposed to 1.7 in controls (p = 0.03).
Studies examining aortic dissection in Turner syndrome have typically involved case reports or small cohorts. In a case report in 1980, only 46 cases of aortic dissection or rupture were published. Matura reports aortic dissection in 3 of 486 women, and mathematically extrapolates these findings to report an incidence of 618/100,000 Turner syndrome patient years.54–56 Boissonnas in 2012 reported 85 total cases of dissection during a time period from 1961 to 2006.57
The average age of aortic dissection is 31 years. This rare but catastrophic event has been associated with hypertension (54% of cases), coarctation of the aorta in 47% and the presence of bicuspid aortic valve in about 27% of cases, although 25% of cases of dissection occur in trileaflet valves without coarctation. Aortic Size Index seems to be 2.5–2.7, although earlier studies report normal ASI. Thus, it remains uncertain which degree of aortic dilatation poses a significant risk for aortic dissection.
A recent review58 suggests an increased risk of aortic dissection with pregnancy and the peripartum period. Although there has been a single case report of maternal death during spontaneous pregnancy, aortic dissection in cases of assisted pregnancy affects 1.25% of cases (1/125 cases); leading to maternal death in 2 of 240 reported cases (0.8% risk). This risk has also been reported59 at 10% of cases, occurring mainly in the third trimester and causing maternal death in 3.5% of cases. This increased risk (up to 100 folds increase) has been postulated to be due to the exacerbation of hypertension by pregnancy, in the context of other cardiovascular defects such as coarctation of the aorta.60–63 However, the true incidence of aortic dissection in relation to fertility remains largely unknown.
Endocrine and metabolic features
Besides affecting only females, the other hallmark of the condition is primary ovarian failure due to insensitivity to pituitary drive. Primary amenorrhea, delayed sexual maturation and infertility are seen in 60–90% of cases. Mosaic cases have a lower incidence of gonadal failure. This is unique to Turner syndrome, as opposed to other so-called “connective tissue disorders” such as Marfan syndrome.
About 16% of girls undergo spontaneous menstruation at an average age of 13 years. Since adrenal function is normal, some of the sexual characteristics do develop; albeit in a reduced form. The lack of gonadal estrogens also negatively impacts skeletal growth, since the normal pubertal increase in estrogen seems to have a beneficial effect counteracting that of premature growth plate fusion due to SHOX haploinsufficiency. Hormonal replacement therapy has become standard practice in many countries (80% in Europe and approaching 70% in the US), usually begun around 12 years of age and not beyond 15.64–68 Although the protective, antiatherogenic role of female sex hormones in the vascular wall has been extensively studied, the effects of spontaneous fertility, pregnancy or female sex hormonal replacement therapy on the structure and composition of the aortic wall in Turner syndrome are still unclear.
Females with Turner syndrome have a distinctly accelerated pattern of the metabolic syndrome, which significantly contributes to their mortality and morbidity risk. Almost half of these patients develop Diabetes mellitus Type–II as early as 5 years of age, mainly due to insulin resistance. Perhaps as a result of this, hypercholesterolemia and hypertriglyceridemia appear as early as 11 years of age. Insulin-like Growth Factor, Insulin-like-Growth Factor Binding Protein and serum cortisol are also elevated. Their lipid profile exhibits a stark pro-atherogenic pattern, quite out-of-proportion to their age. Likely due to the increase in Leptin, the adipose tissue distribution differs significantly from normal controls, with a shift toward an increased visceral and truncal fat and a reduction in the typical “gynecoid” distribution of fat seen in normal females.
Obesity is another major finding; with Body Mass Index and Total Body Fat Mass significantly higher and Lean Body Mass significantly lower than age-matched controls. Several cytokines are increased, including interleukins IL-6, IL-8, C-Reactive Protein, circulating endothelial adhesion molecule ICAM-1 and Tumor Necrosis Factor-α, all markers of a chronic, pro-fibrotic inflammatory response. Some evidence suggests an increased activity of the Renin–Angiotensin–Aldosterone system.47,68–73 There is also speculation about the role of Vascular Adhesion Protein-1, a cytokine implicated in various chronic inflammatory conditions such as diabetes, congestive heart failure and atherosclerosis, all of which are common findings in Turner syndrome.
Remarkably, these changes are independent from the patient’s karyotype or estrogen hormonal replacement therapy.
Discussion
The underlying cause of Turner syndrome remains unclear. While not shown to be due to a distinct genetic or transcriptional mutation or defect, its clinical manifestations clearly demonstrate a picture of developmental and differentiation failure involving the skeletal, ovarian and aortic arch. The findings also show a distinct pattern of a chronic, low-grade inflammatory response characterized by an increased deposition of fibrous tissue, increased aortic wall rigidity and a premature or early onset of the Metabolic syndrome, reflecting a pattern typically seen in ageing.
With regard to cardiovascular risk, the major cardiovascular events remain coronary artery disease, myocardial infarction and stroke; quite in concert with the expected outcomes given the metabolic, endocrine and vascular pathophysiology. Despite the increased incidence of bicuspid aortic valve morphology, this does not translate into an increased risk of aortic dissection. Several studies have shown that bicuspid aortic valve alone is not a risk factor for aortic dissection74 (Thoralf Sundt, MD. Personal communication. July 10th, 2014). In addition, the modest aortic dilatation seen in Turner Syndrome is associated with vascular wall changes similar to those seen in atherosclerotic disease, and almost diametrically opposite from those seen in Marfan syndrome. Specifically, the aortic wall is thickened, with normal and even increased collagen and fibrous tissue content, leading to an increased rigidity and decreased compliance (flexibility or distensibility) of the vessel.
The effects of spontaneous fertility, assisted fertility, female sex hormone therapy, oral contraceptives, pregnancy and peripartum period on the vascular wall are still unclear. As such, the aorta has an increased structural strength and more likely to withstand the force of pulsatile stretch, which is again the opposite of Marfan syndrome.
The current body of evidence clearly demonstrates that Turner syndrome should not be included under the blanket terms “connective tissue disorders” or “genetically mediated diseases” since it is primarily a developmental disorder and the impact of chromosomal and genetic abnormalities is still incompletely understood.
Moreover, the cardiovascular lesions and manifestations associated with Turner Syndrome are still incompletely understood. However, current evidence suggests that the increased risk of aortic dissection and assisted fertility, pregnancy, the puerperium and hypertensive emergencies in these patients, which has led Dr Bondy to recommend against pregnancy in this population.81–85 This has led to an increased focus on management of hypertension in a younger age to help mitigate the associated risk of acute aortic syndromes. However, there are no data or comprehensive guidelines for antihypertensive management in these patients, which are still based on “expert opinion”, even though standard therapies (such as β-blockers or calcium channel blockers) have limited or even harmful effects.35,75–80 This is particularly important in pregnancy, where hypertension leads to pre-eclampsia in 20% of cases and severely increases the risk of death.
Conclusions
Ever since the early reports86,87 in 1930 by Otto Ullrich and 1938 by Henry Turner, a diagnosis of Turner syndrome has had a life-changing and everlasting impact on the affected female and her family. Affected girls and women face numerous challenges in multiple aspects of their lives; developmentally, biologically, socially and career-wise. Their self-image and self-esteem is severely impacted by the clinical manifestations, including feeling “less feminine” because of their short stature and delayed or absent sexual maturation, as well as the associated visual-spatial and behavioral changes. Given the continuous struggles to cope with such adversities, it is not surprising to see such females retiring at the relatively early age of 40 or 45, even among those who have earned advanced higher education.88–95
Adding to the frustration of dealing with this multi-faceted chronic disease is the unclear recommendation relating to management of the cardiovascular risk. Current guidelines are based on an incomplete set of low-level evidence, but they continue to demand a high-level intervention that has a serious impact on the life of every female with Turner syndrome. Given that clinical practice guidelines are intended to provide diagnostic and therapeutic interventions whose risk is far less than that of the disease process, the recommended invasive surgical interventions in an asymptomatic patient may need to be revised with more focus on identifying and addressing the risk factors for life-threatening acute aortic events. Control of hypertension, counseling regarding reproductive function and a multi-disciplinary approach offer potential benefit in management.
More extensive, population and registry-based as well as basic science and translational studies are urgently needed to provide the necessary scientific basis for correctly estimating the mortality and morbidity risks among this specific group of patients.
Conflict of interest statement
The author holds a full United States patent for a Total Aortic Arch Reconstruction Graft. No financial relationship to disclose. Otherwise, there is no conflict of interest.
Acknowledgements
The author is deeply grateful and appreciative for all the invaluable insights from girls and women with Turner syndrome, the Turner Syndrome Societies in the US, Canada and the UK and all the dedicated professionals caring for these patients. The author is also deeply grateful to Ms Nicola Wood, librarian at the Royal Society of Medicine, London, UK for her assistance with the literature search.
This study has not received any funding from any organization or government.
References
Cite this article
TY - JOUR AU - Hisham M.F Sherif PY - 2016 DA - 2016/08/04 TI - Turner syndrome and guidelines for management of thoracic aortic disease: Appropriateness and utility JO - Artery Research SP - 21 EP - 29 VL - 15 IS - C SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2016.07.001 DO - 10.1016/j.artres.2016.07.001 ID - Sherif2016 ER -