
Growth Hormone Therapy for Paediatric Growth Disorders: The Past, Present, and Future
- DOI
- 10.2991/dsahmj.k.200227.001How to use a DOI?
- Keywords
- Growth hormone therapy; pituitary; efficacy; safety
- Abstract
Abnormal pituitary function was linked to excessive height and acromegaly in the late 19th century. Harvey Cushing proposed a pituitary substance that regulated human linear growth. Chemical characterization of the Growth Hormone (GH) molecule by Li led to a demonstration of its species specificity with only Human GH (hGH) stimulating growth in children. Extraction of hGH from cadaver pituitaries and its purification led to therapy by Raben in the 1950s, demonstrating growth acceleration in children with hypopituitarism. From the 1960s until 1985, pituitary-extracted hGH was used as therapy in GH-deficient children. In 1985, an epidemic of Creutzfeldt–Jakob disease, caused by contamination of pituitary-derived hGH with the prion protein, caused the deaths of more than 200 children worldwide. Cadaveric hGH was discontinued and replaced by Recombinant hGH (rhGH), which was licensed by the US Food and Drug Administration and European Medicines Agency for GH Deficiency (GHD), followed by non-GHD disorders, Turner syndrome, small for gestational age short stature, and idiopathic short stature. A mathematical model predicted human growth responses, but despite rhGH’s wide use and availability and good safety record, long-term responses remain variable. Poor adherence to rhGH has led to development of weekly or fortnightly hGH administration using different techniques to prolong activity. Data on tolerability, efficacy, and long-term safety of long-acting rhGH preparations will make up the next stage of the development of this important hormone therapy.
- Copyright
- © 2020 Dr. Sulaiman Al Habib Medical Group. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
It is now more than 60 years since treatment with Human Growth Hormone (hGH) was first demonstrated to stimulate linear growth in children with hypopituitarism lacking endogenous GH secretion. As we are now moving well into the 21st century, it is appropriate to appraise the progress of this therapy, which has gone through several important stages of development and is still evolving.
Abnormalities of human linear growth such as so-called “dwarfism” or excess height were brought to the attention of the public in the 19th century and exhibited for financial exploitation and gain. There was no concept at that time to rectify or correct the physiological mechanism leading to the outward signs of clinical abnormality. It was the recognition of the association of the human pituitary gland with physical growth disturbance, for example, clinical overgrowth features were linked to an enlarged pituitary gland at autopsy [1], which pushed knowledge forward in the direction of the understanding of human pathology. The term “acromegaly” was coined in 1886 [2], and the presence of a pituitary tumour was subsequently reported to cause both short stature and obesity, suggesting that pituitary disturbance could also result in suppression of growth [3]. It was Dr. Harvey Cushing, the famous pioneer of brain surgery, who while based at Johns Hopkins Hospital in Baltimore, first considered that a hormone secreted by the pituitary gland could be involved in growth regulation [4]. Hence, the concept of a growth hormone was born, launching the process of a search for the identities of pituitary hormones and their functions.
The chemical structure of a putative pituitary growth hormone was intensively studied by Dr. H.M. Evans and his team at the University of California, San Francisco, and the first pituitary extracts were administered as therapy to rats in 1921, leading to a measurable effect of bovine GH on growth promotion [5]. Subsequently in the same laboratory, the chemical structure of hGH was identified by Dr. Li working closely with Evans [6], as a protein with 191 amino acids and two disulfide bonds.
The opportunities for using pituitary-extracted GH clinically were then investigated, and growth promotion in hypophysectomized monkeys was demonstrated after administration of pituitary-extracted GH. Similar data were reported in humans, but Ernst Knobil, who demonstrated the species specificity of hGH, while working at Harvard [7]. This knowledge was instrumental in the establishment of a National Pituitary Agency in the United States with the aim of collecting human cadaver pituitary glands from which GH was purified for treatment of so-called “dwarfism” in paediatric GH deficiency. Extraction of GH from human pituitaries also became a widespread practice throughout Europe. The first clinical description of successful growth promotion using extracted pituitary hGH was given by Dr. Maurice Rabin, who was working at the Tufts Medical Center in Massachusetts in 1958 and subsequently in 1962 [8,9]. Key milestones in the story of GH therapy are shown in Figure 1.
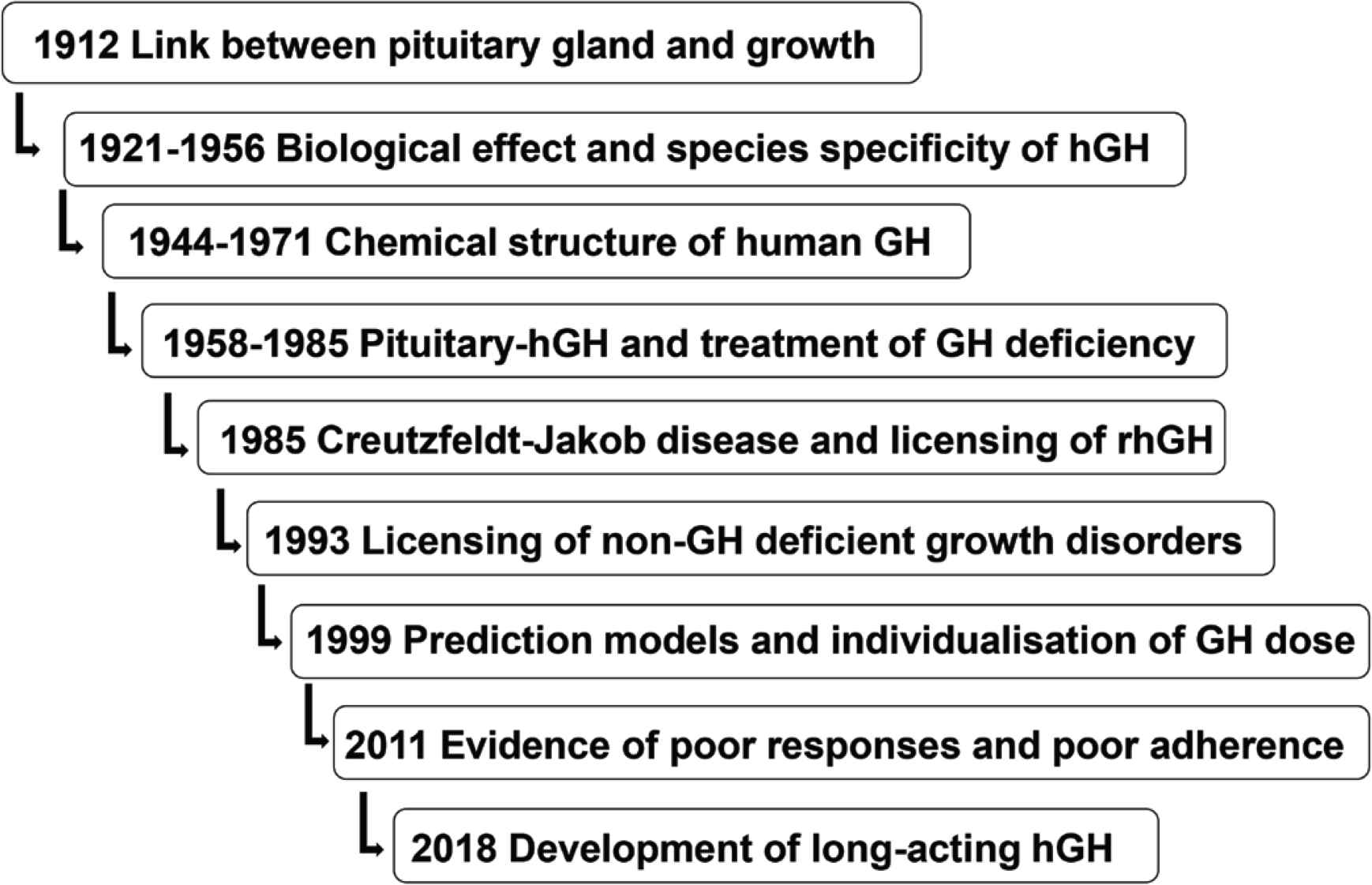
Key milestones in the development of GH therapy over more than 60 years [13]
2. THE ERA OF PITUITARY-EXTRACTED hGH
From 1963 to 1985, national systems were put in place in many countries for the collection, extraction, and purification of pituitary hGH. Although efficacy in terms of growth promotion had been clearly demonstrated, supplies remained scarce and were strictly limited to treatment of severe GH deficiency. The pharmaceutical industry contributed by establishing commercial techniques of hGH purification that were free of contamination by other pituitary hormones.
2.1. Progress in Diagnosis of GH Deficiency
Progress was being made in parallel on the diagnosis of GH deficiency in children. The first immunoassay for determination of serum GH was published by Glick et al. [10] in 1963, and this assay was used to quantitate the serum GH response to insulin-induced hypoglycemia [11], which remains the gold standard test for the diagnosis of GH deficiency, although it is seldom used in children because of the risk of severe hypoglycemia. In the late 1960s, a peak stimulated GH level of 5 μg/L was considered the cutoff level for exclusion of G deficiency; however, the appreciation of a range in severity of paediatric GH deficiency led to a relaxation of this definition, with many clinicians using a cutoff of 10 μg/L. Only recently has a cutoff value of below 7 μg/L been more widely accepted as the international definition of GH deficiency. Over the years, the international reference preparation (IRP) for the hGH assay depended initially on purified pituitary GH and then on purified 22 kDa recombinant hGH with a potency of 3 IU/mg [12]. Today, a serum concentration of 20 μIU hGH is equivalent to 6.7 ng [13].
The discovery of the control of GH secretion through a balance between the two hypothalamic releasing peptides somatostatin and GHRH, characterised in 1973 and 1982, respectively [13], resulted in demonstration of pulsatile GH secretion in children [14,15]. However, in clinical practice measurement of multiple samples was not a convenient method for clinical investigation; therefore, pharmacological stimulation tests became the established procedure for diagnosis. The range of diagnostic tests increased to include stimuli such as arginine, l-DOPA, glucagon, and propranolol [16].
It became appreciated that the diagnosis of GH deficiency in children was a multifaceted process requiring a combination of clinical assessment and tests of the GH–Insulin-like Growth Factor 1 (IGF-1) axis investigation and brain Magnetic Resonance Imaging (MRI) [12]. The MRI criteria for severe GH deficiency are ectopic posterior pituitary gland, an indistinct pituitary stalk, and a small volume pituitary gland consistent with pituitary hypoplasia [17].
2.2. Treatment with Pituitary-extracted hGH
The GH-deficient patients treated with pituitary hGH were usually rather old, that is, >10 years, with severe short stature and usually had multiple pituitary hormone deficiencies. The hGH was administered two or three times weekly by intramuscular injection, and it was only in 1983 that daily injections were shown to be more efficacious [18] and that subcutaneous administration was equally effective. The dose of pituitary hGH was gradually modified from 10–20 IU/week to a dose of 30–100 mIU/kg body weight given three times weekly, which gave superior responses [19].
2.3. Creutzfeldt–Jakob Disease
In 1984, Dr. Ray Hintz reported the first prismatic case of the serious neurodegenerative condition known as Creutzfeldt–Jakob Disease (CJD) in a 20-year-old patient who had been treated with cadaveric pituitary hGH for 14 years [20]. The patient died within 6 months of the onset of symptoms. Post-mortem examination showed the spongiform encephalopathy characteristic of this condition and caused by misfolding of a Prion Protein (PrP). The disease is caused by infection with a pathological prion protein (PrPSc), which leads to death of brain tissue. If the abnormal prion had its origin in an infected cadaver pituitary gland and escaped the purification process, the infective agent could be passed to patients receiving pituitary hGH. A global epidemic of CJD ensued with a total of 226 cases being reported in patients who had received pituitary hGH. Prescription of cadaver-extracted hGH immediately ceased in virtually every country. The chapter of pituitary-derived hGH was over (Figure 1).
2.4. Recombinant hGH
In the 1970s, genetic engineering technology enabled the expression of the DNA sequence encoding recombinant hGH (rhGH) [21]. The peptide initially contained an additional methionine residue (meth-hGH) and in 1985, the first hGH containing methionine produced from Escherichia coli was approved by the Food and Drug Administration (FDA) for treatment of GH deficiency in children (Figure 2). Safety and efficacy were documented in obligatory postmarketing surveillance studies, organized by pharmaceutical companies, and formed the extensive databases of the Kabi International Growth Study in Europe and the National Cooperative Growth Study in the United States. Both have provided a wealth of important scientific information. With the restriction on supplies of hGH now removed, GH therapy moved into a new era, with a much less restrictive approach adopted by clinicians. Children diagnosed as GH deficient were generally younger and had milder degrees of GH deficiency. An hGH regimen of 33 μg/kg body weight/day was generally adopted.
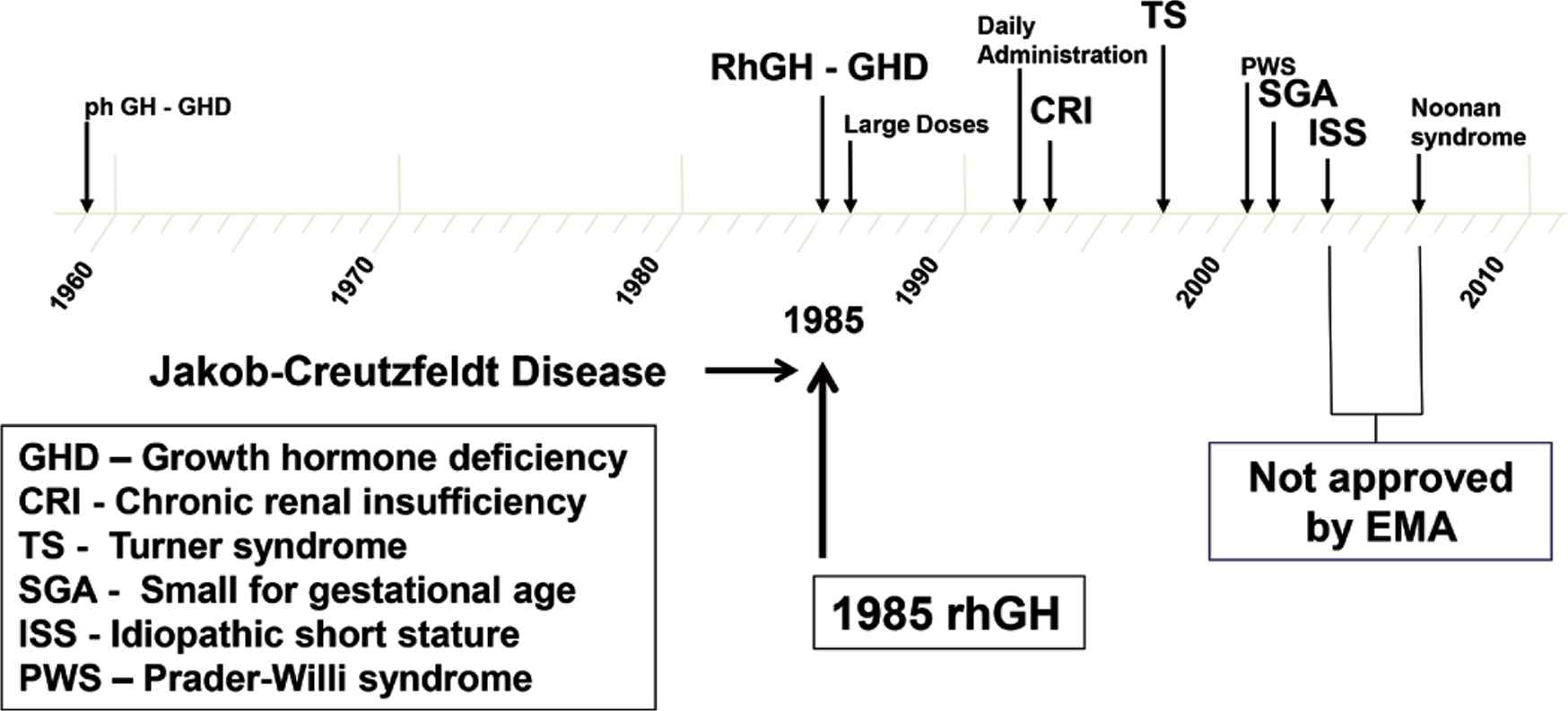
Growth disorders with approval for growth hormone therapy from the United States Food and Drug Administration and European Medicines Agency (EMA)
2.5. Treatment of Non-GH Deficiency Disorders
Trials with rhGH were started in non-GH deficient disorders such as Turner syndrome, short stature in children born Small for gestational age (SGA), Idiopathic Short Stature (ISS), and more recently SHOX gene deficiency (Figure 2). A randomized controlled clinical trial in Turner syndrome showed significant improvement in final height in rhGH-treated patients compared with untreated controls. Early onset of treatment using an hGH dose of 50 μg/kg/day was recommended [22]. The FDA and the European Medicines Agency (EMA) approved Turner syndrome for rhGH therapy in 1996. Growth failure in chronic renal insufficiency had previously been approved in 1963.
Short stature related to SGA became a further approved indication, by the FDA in 2001 and the EMA in 2003. Treatment with rhGH had demonstrated gain of adult height provided that hGH therapy was initiated more than 2 years prior to the start of puberty [23]. Although the efficacy of rhGH in inducing adult height gain was now demonstrated by a number of groups, there was debate regarding the optimal hGH dose. The group from Rotterdam proposed 33 μg/kg/day [24], but other groups maintained that a higher dose of 67 μg/kg/day induced better quality catch-up growth [25]. The adult height data submitted to the EMA for registration of rhGH therapy indicated that both doses had similar effects on adult height; however, the higher dose tended to induce supraphysiological levels of serum IGF-1 during therapy. Hence, the lower dose of 33 μg/kg/day is now recommended by the EMA [26,27].
Abnormal growth related to so-called “idiopathic short stature” posed a further challenge. ISS can be defined as height <1.25 height centile and predicted adult height of <160 cm in males and <150 cm in females. The category of patients known as ISS is generally considered to embrace a wide range of etiologies associated with normal endocrine status, absence of a chromosomal or dysmorphic syndrome, and normal birth weight. In the United States, a randomized, placebo-controlled study demonstrated superior growth during rhGH therapy [28], which together with other randomized studies led to ISS being approved by the FDA as an indication for rhGH [29]. The EMA, however, rejected the application for ISS on the grounds of lack of proven efficacy. Nevertheless, it is appreciated that many children carrying the label of ISS will benefit from long-term rhGH therapy, although the quality of growth response varies markedly between groups. It was generally recommended that if rhGH therapy is to be attempted, a dose of 50 μg/kg/day should be used [29]. More recent approvals for rhGH therapy have included SHOX deficiency [30] and Prader–Willi syndrome, largely on the basis of improved muscle strength, physical activity, and body composition [31], and Noonan syndrome [32], approved only by the FDA (Figure 2).
2.6. Safety of rhGH
Recombinant human growth hormone therapy has proved to be remarkably safe. Abnormalities reported during therapy tend to reflect the nature of the primary disorder being treated. For example, benign intracranial hypertension and slipped femoral epiphyses may occur in children with severe forms of GH deficiency. There are no clinical data demonstrating that a higher risk of cancer exists in children without a built-in inherent risk. Postsurveillance registries, by recording adverse drug reactions, have contributed significantly to the overall positive reputation of rhGH as regards safety. The frequency of adverse events is lowest in patients with idiopathic GH deficiency or ISS [13]. EMA recommendations regarding dose of rhGH for each indication need to be adhered to, and when prescribed according to these recommendations rhGH therapy has so far appeared to be very safe [33].
2.7. The Continuum Model of GH–IGF-1 Axis Defects
The continuum model of GH–IGF-1 axis defects [34] is relevant to the story of rhGH therapy (Figure 3). The function of this axis may be disturbed by defects occurring at a number of sites. If one takes the analogy of a flowing river stream, the head of the river equates with the upper end of the axis starting with factors controlling secretion of GH. After a series of complex interactions, the mouth of the river can be equated to the delivery and action of IGF-I. Consequently, the axis extends from defects of GH secretion, starting with severe GH deficiency, and moving through milder defects to ISS and then into defects of GH action, namely, GH resistance abnormalities, which can also vary from mild to severe [35]. GH–IGF axis defects, therefore, make up a continuum from GH deficiency to GH resistance [34] (Figure 3).
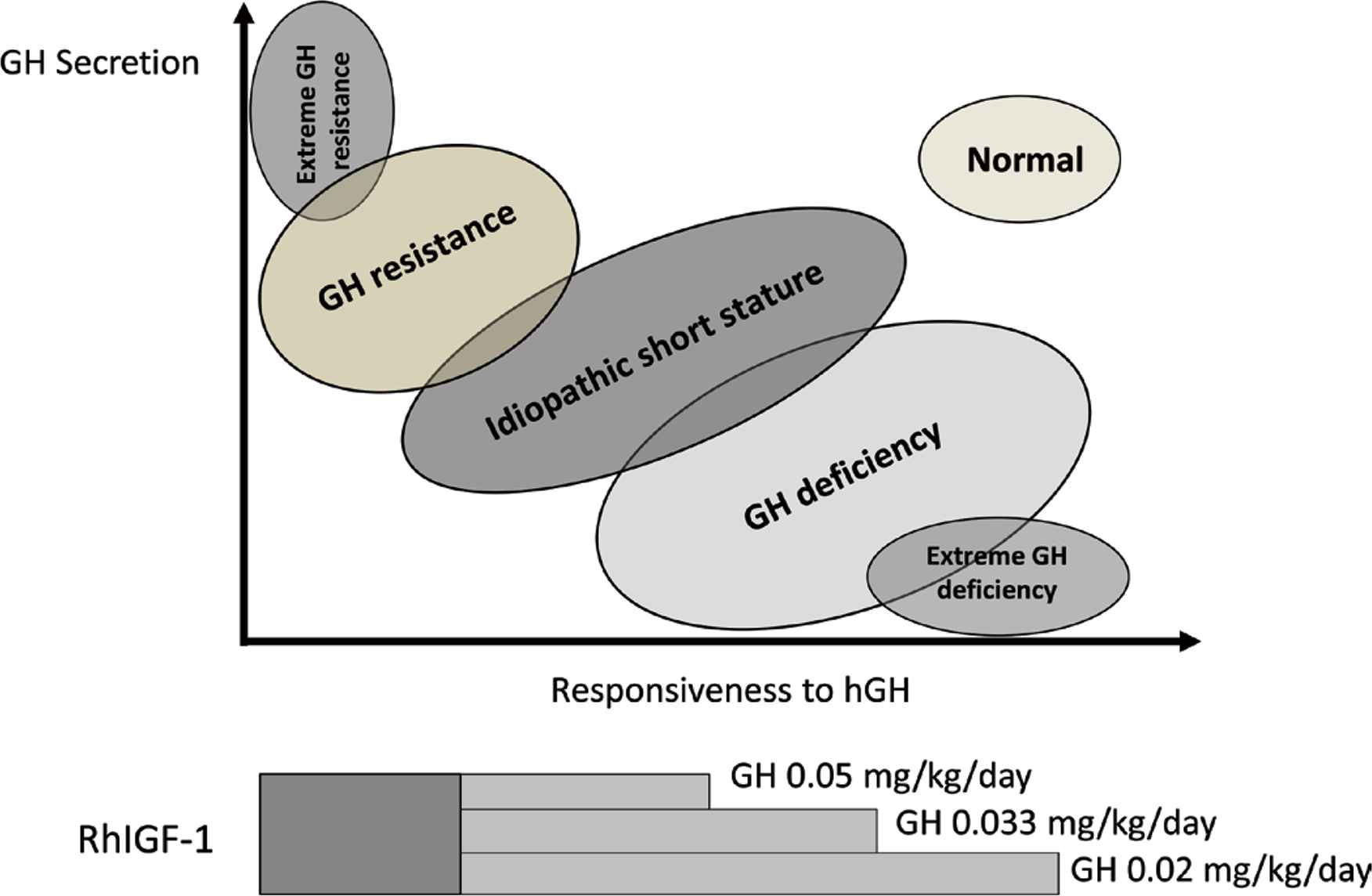
The continuum model of GH–IGF-1 axis defects showing recommended treatment with rhGH or rhIGF-1 [34]
These abnormalities present clinically with short stature, and the clinician has a responsibility to address this challenge with therapy that is safe and has had demonstrated efficacy. The two ends of the continuum—GH deficiency and GH resistance—can be treated with licensed rhGH and rhIGF-1, respectively [36]. It is the intermediate defects that are more challenging. As we will discuss below in the context of growth prediction models, severe GH deficiency is more responsive to rhGH than mild GH deficiency; consequently, a high rhGH dose of ~35 μg/kg/day would be more appropriate to induce an expected growth response, as shown in Figure 3. As mentioned above, the patient with ISS requires an even higher dose, as this patient is situated further down the X-axis of GH responsiveness in the continuum model. When patients with GH resistance, also known as primary IGF-1 deficiency, are encountered, the clinician must appreciate that they will be unresponsive to rhGH. A tendency to try these patients on higher doses of rhGH must be resisted, and the best option for their management is replacement with rhIGF-1, which is the logical approach as their primary defect is causing a deficiency in IGF-1 that is not responsive to rhGH [35].
2.8. Growth Prediction Models
Despite the major therapeutic advance that use of rhGH represents, there remains considerable variability of long-term growth response, with many patients not achieving their genetic potential, even in classical GH deficiency [37]. To address this variability, a range of models can predict the growth response to GH therapy given for different indications [38–42]. Working from such models, height velocity using a standard dose of rhGH 0.3 mg/kg/week will increase by 2 cm/year in children with GH deficiency, compared with SGA and Turner syndrome patients. The highest rated predictor for children with GH deficiency is severity of the condition compared with the dose of hGH per kg body weight per week in patients with SGA and Turner syndrome [43].
Given the mathematical complexity of these models, which have been shown to reduce the number of poor responders to rhGH [44], the practical uptake of the models in the consultation setting has been disappointing. However, understanding and using the different variables, particularly when calculating the initial dose of rhGH, is strongly recommended.
2.9. Poor Adherence to rhGH Therapy
The difference between observed and predicted gain in height or height velocity can be a measure of the adherence to the hGH treatment regimen. If responsive reduced while the diagnosis is correct and the presence of intercurrent disease is ruled out, poor adherence to the hGH regimen should be considered [45,46]. Monitoring of adherence should begin as the hGH treatment is initiated because some patients may not be compliant from the start or not renew their prescriptions, particularly if the evidence of real benefit is lacking [47–49]. Measurements of IGF-1 SDS may give an indication of adherence with GH, but this is often not determined routinely and may not provide a definitive answer [50]. Poor adherence with hGH has been associated with unsatisfactory clinical outcomes [51]. Assessment of adherence has generally been poor, but have indicated that up to 82% of patients may miss at least some doses of GH [49].
Effective management of poor adherence to hGH requires the paediatric endocrinologist or specialist nurse to learn techniques of nonjudgmental motivational interviewing [52]. Organization, time, knowledge of common issues affecting adherence, and the ability to build a close relationship with the family with open questions and emphasis on pre-hGH treatment education are key components of this type of HCP–patient interaction. The same healthcare professional should discuss adherence at each outpatient visit. The process also involves addressing the choice of hGH brand and hGH injection device. Updated electronic monitoring is improving this process and provides important feedback data on evidence of suboptimal adherence, which may not be available from self-reported data from patients and caregivers, clinical history, or auxological measurements.
Electronic monitoring of GH injections can be performed using the easypod injection device and Easypod Connect system, which is the only such device currently approved [45]. The device facilitates administration of a preset dose of hGH, records injection times and doses, and provides the patient with information such as number of doses remaining [53]. The injection information can be collected and downloaded via Easypod Connect by healthcare personnel, which enables distance monitoring, with less need for frequent hospital visits. Thus, healthcare personnel can address issues of nonadherence with treatment at an early stage prior to any decrease in GH efficacy. Studies to date have indicated good tolerability of the device and high levels of adherence over several years [54].
2.10. Long-acting rhGH Preparations
Over the course of more than 60 years since hGH therapy was first introduced, there have been many “casualties” of alternative therapeutic approaches for achieving the biological effect of administered rhGH. Notably, these have been oral and intranasal forms of hGH and GH secretagogues or GHRH aimed to stimulate endogenous pituitary GH release. In many fields of medicine, long-acting preparations have replaced daily injections, and it appears that a new era of long-acting hGH preparations is about to dawn. There are three basic approaches to prolong the action of GH: (1) GH pegylation, (2) attachment of the GH molecule to albumin or amino acid tails, and (3) linking an inert peptide with the rhGH so that the unaltered hGH molecule is released over a prolonged period. Several pharmaceutical companies have reported encouraging data on noninferiority of growth response in GH-deficient children compared with daily rhGH therapy in controlled phase II and III studies [55,56].
Long-acting rhGH preparations, providing weekly or fortnightly administration, may represent an advance because of increased convenience and improved adherence and growth responses. However, long-term surveillance will be necessary with detailed assessment of efficacy, safety, cost–benefit, and quality of life. It is likely that long-acting rhGH preparations will have an impact on hGH replacement therapy; however, the size of this impact is currently unknown [57]. Tolerability in terms of injection pain, concentrations of serum IGF-1 during treatment, and of course the cost of the therapy are likely to dictate the acceptability and uptake of this new form of rhGH therapy.
3. CONCLUSION
The story of hGH therapy in children started with the recognition of the pituitary gland as the driver of linear growth and reaches a point, more than 60 years later, with a technological discussion of new genetically engineered hGH preparations. RhGH administration has changed the lives of children with true GH deficiency by inducing catch-up and long-term height gain, often into the normal range. In non-GH deficient disorders, a measured impact has been seen, but this cannot be described as ideal because the effect of the primary condition generally persists to influence both height and phenotype. The new era of long-acting rhGH will not revolutionise therapy, but may provide a refinement and a possible degree of finesse to the story of the development of this important growth-promoting therapy.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
MOS and AA contributed to the writing of the manuscript.
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - Martin O. Savage AU - Abdullah Alherbish PY - 2020 DA - 2020/03/03 TI - Growth Hormone Therapy for Paediatric Growth Disorders: The Past, Present, and Future JO - Dr. Sulaiman Al Habib Medical Journal SP - 4 EP - 9 VL - 2 IS - 1 SN - 2590-3349 UR - https://doi.org/10.2991/dsahmj.k.200227.001 DO - 10.2991/dsahmj.k.200227.001 ID - Savage2020 ER -