
EGFR Microdeletion Mutations Analysis System Model Using Parameters Combinations Generator for Design of RADBAS Neural Network Knowledge Based Identifier
- DOI
- 10.2991/ijcis.11.1.93How to use a DOI?
- Keywords
- RADBAS Neural Networks; knowledge base identifier; EGFR gene; microdeletion mutations
- Abstract
The aim of this research is to automate an analysis of the EGFR gene as a whole, and especially an analysis of those exons with clinically identified microdeletion mutations which are recorded with non-mutated nucleotides in a long chains of a, c, t, g nucleotides, and “-“ (microdeletion) in the NCBI database or other sites. In addition, the developed system can analyze data resulting from EGFR gene DNA sequencing or DNA extraction for a new patient and identify regions potential microdeletion mutations that clinicians need to develop new treatments.
Classifiers, trained using limited set of known mutated samples, are not capable of exact identification of mutations and their distribution within the sample, especially for previously unknown mutations. Consequently, results obtained by classification, are not reliable to select therapy in personalized medicine. Personalized medicine demands exact therapy, which can be designated only if all combinations of EGFR gene exon mutations are known.
We propose computing system/model based on two modules: The first module includes training of knowledge based radial basis (RADBAS) neural network using training set generated with combinatorial microdeletion mutations generator. The second module has two modes of operation: the first mode is offline simulation including testing of the RADBAS neural network with randomly generated microdeletion mutations on exons 18th, 19th, and 20th; and the second mode is intended for application in real time using sample patients’ data with microdeletion mutations extracted online from EGFR mutation database. Both modes include preprocessing of data (extraction, encoding, and masking), identification of distributed mutations (RBNN encoding, counting of exon mutations distribution and counting of EGFR gene mutation distribution), and standard reporting. The system has been implemented in MATLAB/SIMULINK environment.
- Copyright
- © 2018, the Authors. Published by Atlantis Press.
- Open Access
- This is an open access article under the CC BY-NC license (http://creativecommons.org/licences/by-nc/4.0/).
1. Introduction
Cancer research is currently one of the leading fields of clinical research. Pulmonary malignances including Non-Small Cell Lung Cancer (NSCLC) are the most common cancers worldwide and the leading cause of death [1–4]. Lung cancers are classified according to histological type, and the vast majority of lung cancers are carcinomas, malignancies that arise from epithelial cells [5]. The two broad classes of lung cancers are Small-Cell Lung Carcinoma (SCLC) [6] and NSCLC. A potential treatment target for NSCLC is the Epidermal Growth Factor Receptor (EGFR) and associated genes. Different combinations of mutations within the gene coding for EGFR exist in cancers of patients with NSCLC. The most frequently observed microdeletion mutations are on the exons 18, 19, 20, and nucleotide conversion on exon 21[7]. Experienced pathologists perform identification and molecular classification of lung cancers performed by utilizing lung biopsies; however, well-equipped laboratories and experienced pathologists are rare and costly, and reliable pathological diagnosis is not always available. Computer aided simulations are a tool through which diagnostics and treatment of NSCLC could be explored.
Further, due to developed computer models clinicians no longer need to analyze long nucleotide gene chains, for example EGFR gene contains 193307 nucleotides [8]. The gene analysis is primarily subjective process depending on clinicians’ competences in informatics. It would be very beneficial to replace the process with a more objective alternative: a tool that can provide clinicians with precise information about EGFR gene status, and of mutated nucleotides distribution, in a short time. If detected gene mutations were statistically reliable, targeted treatment for a patient can be started. If detected gene mutations are positioned in new mutation regions of specified exons, clinicians and pharmacists need to discover a new potential targeted treatment. Treatments targeting known nucleotide mutations, if used to treat new gene mutations would expose patients to unnecessary side effects.
Background, scientific sources and publications referenced in this paper can be roughly classified into five categories: Artificial Neural Networks (ANN) as a decision-making tool in the field of cancer, ANN in detection of cancer cells or nodules, ANN in classification and prediction of mutated genes-exons, identification of nucleotides gene mutation, and personalized medicine.
The advantages of using the ANN as a supportive tool for decision making processes in cancer diagnosis and treatment has been previously described by Paulo J. Lisboa et al. [9]. In this publication, the authors explain areas where the method of neural networks give best results and provide basic algorithms.
K.A.G. Udeshani et al. [10] focused to accuracy of nodule detection using pixel and statistical texture features as the inputs to train a neural network. Zhi-Hua Zhou et al. [11] created a system based on image processing and several joined neural networks for automatic cell cancer diagnosis with a focus to improvement of detection reliability using voting system based on Neural Network Ensemble. A step towards the clinical validation in detection of lung nodules has been made using the metric of sensitivity (reduction of false positives) in the approaches based on classifiers (Support Vector Machines-SVM and Extreme Learning Machine-ELM) [12] and massive training ANN[13].
In their publication, Emmanuel Aetiba et al. [14] tried to find optimal backpropagation algorithm for networks training for NSCLC diagnosis and tested several algorithms. Considering that the deletions on exon 19 are the most common mutations of the EGFR, a nucleotide sequence of the exon 19 was used for neural networks training. The best performance with the minimal number of epochs and training time were accomplished by using the Levenberg-Marquardt algorithm. Aetiba et al. also [15] used the EGFR nucleotide sequence with microdeletion mutations for different neural networks training and described two new algorithms called SimMicrodel and GNEN. The SimMicrodel was used for the simulation of statistical mutations for different categories of patients with microdeletions for exons 18, 19 and 20. The GNEN was used for encoding exons TK domain for normal and mutated genes. Different neural networks were built using simulated samples of the genome within all of the algorithms.
Modern methods are increasingly turning to the identification of mutations or the total sequencing of genes. The paper [16] identified EGFR exon 20 insertions through an algorithmic screen of 1,500 lung adeno-carcinomas.
The discovery of EGFR advanced our understanding of the molecular basis of lung cancer. These findings encouraged further investigations of EGFR-TKI mutations and their role in predicting drug sensitivity in the field of personalized medicine. Clinical response to targeted therapy based on identified mutated nucleotides of 18–21 exons is a key indicator of the effectiveness of a given anticancer treatment [16].
The papers [10, 11, 12, 13] discuss lung cancer at the molecular level (or cancer cell detection, or cancer nodule detection. It remains unknown which gene is mutated, which exons are mutated, which is a type of mutation and what the structure of the mutated gene region (exons, position and number of mutations). Approaches [10, 11, 12, 13, 14, 15] focus on the accuracy of the classifier training and testing, but clinical acceptability of a novel approach, method or tool requires are liable sensitivity and specificity value metrics. A study by [14] explored the approximate evaluation of statistically known mutations only on one exon - 19. The algorithm only performs approximate categorization of mutated exons (patients) in terms of the number of statistically known mutations, but not in terms of positions of mutations within specific exons.
In general, the accuracy of classification of an unknown sample is limited by training sets of samples and sample’s distribution homogeneity on which classifier is based on. Classifiers given a small sample of statistically known different sample data for training [14, 15], perform with lower precision when presented with a set of unknown data, and the accuracy is lower decreasing in relation to the difference between the unknown data and statistically known data.
Multi-Layer Perceptron Network (MLPN) is the most popular technique for classification, and it employs iterative process for training. In our previous research [17], we have used all patients from paper [15] in the training/learning process of the backpropagation neural network. That set of samples covers all statistically mutated sequences, but adds the appropriate number of healthy sequences, which are necessary for the proper training. In this set, we have replication of patients with the same kind of mutations. When such set was brought to our algorithm, it was divided into training pairs (70%), check validating pairs (15%), and test pairs (15%). A certain number of validating and test vectors (or all validating and test vectors) were the same as the training vectors. As such, the validating and test errors are very small and close to the training error. In addition, regression plots for training, validation, and test were close to 1.These results are not objective and reliable when new mutated exons (which are not part of the initial training set) were introduced to our backpropagation neural network. Consequently, we continued to improve the accuracy of our solutions and we integrated all unique 30 samples in one matrix. Best-obtained networks had very poor results (training error = 0.001, validation error = 5.94, regression training R=0.8461, regression validation R=0.38099, and regression test R=0.18265).
In the process of external validation, this approach gave us approximate values of classes for the statistically known mutated exons, but for newly mutated exons, we had unreliable (unusable) values. Such values may not be acceptable in personalized medicine as the exact position and number of mutated nucleotides in exons cannot be determined.
However, using MLPN for a classification of samples with different mutated nucleotides into the same class does not provide precise information for selecting the right treatment. Several biomedical research papers [18, 19, 20, 21, 22, 23, 24, 25] report results of using the RBNN as a classifier. These classifiers performance is linked to approximate values, and consequently advising on approximate treatments and such approach is not suitable for personalized medicine.
In this context, the application of RBNN for mapping one input to one output resolves an approximate classification of mutant exons into exact deterministic identification of the positions of mutated nucleotides within the exon. RBNN is designed in a single iteration and learn applications quickly. To identify each individual vector, the most common method is using RBNN to assign a single neuron for each input [19]. This would not be practical for all vectors created by a binary combinations within an exon due to the dimensionality of the exon. Because of that, we concluded that new approach based on binary combinations of parts of an exon with dimensionality that introduces a satisfactory number of neurons would be the right solution.
For an exact treatment to be utilized by clinicians a new approach is needed for identification of mutated exons based on structural information of mutations on exons. Authors in [26] turn to the identification approach for mutated exon 20 in lung adeno-carcinomas, but gene 20 is analyzed and experimented only from the aspect of insertions-mutations.
Motivated with the deficiency identified in previously described solutions from literature, and based on our own experience, we have decided to replace the approach of using a classifier for approximate solution with the identification of microdeletions on exons and integration into complete EGFR gene.
The paper is organized as follows: In the introduction, we presented the domain problem and motivation. In section 2 we introduce two approaches for the analysis of mutated exons: (1) statistical oriented exons’ mutations approach resulting in a training set for approximate classifier, and (2) “predictive” oriented exons’ mutations approach resulting in a training set for exact identification. In section 3, the computing system model for EGFR microdeletion mutation analysis using a RBNN [19], knowledge based identifier is presented. Section 3 describes RBNN design and training, and afterwards the simulation of the system for identification of microdeletion nucleotide mutations in EGFR gene exons 18, 19 and 20. In section 4, we discuss validity of our approach and benefits of applying RBNN identifier in our solution. In conclusion, we talk about the purpose of this solution in our long-term research project focused on treatment for lung cancer in relation to biomarkers [19, 33].
2. Materials and methods
2.1. EGFR mutations
A mutation results from an amino acid sequence change in the DNA molecule. The addition or deletion of nucleotides may have a profound effect on the downstream polypeptide. A point mutation is a result of an interchange of one nucleotide. A frame-shift mutation occurs when the frame of the gene that is to be translated is displaced due to an addition or a deletion of a nucleotide [27].
The tyrosine kinase (TK) domain is a region on the EGFR gene, which is prone to mutation in patients with NSLC. The TK domain has 7 exons (exons 18–24), of which exons 18–21 carry somatic mutations in patients with NSCLC [27].
The data about EEGFR nucleotide sequences of healthy exons (18, 19, 20) was taken from NCBI_NG_007726 [8], Fig 1.

Extracted healthy exons 18, 19 and 20 from the full EGFR gene nucleotides sequence
These exon nucleotides sequence were used in our algorithm with the aim to define length of exons and number of parts consisting of ten nucleotides of a particular exon in the process of exploitation of system. The data about microdeletions on the EGFR gene (exons 18, 19 and 20) was taken from the publication [15].
These mutated exons were used in the validation of our model in the exploitation of our system. Correlation between relative positions of nucleotides in exons and absolute position of nucleotides in EGFR gene, as well as microdeletions in exons of statistically known patients are given in Table 1.
| Patient class | Start | End | Positions of mutations (from–to) | Number of patients | Exons | Mutations | |
|---|---|---|---|---|---|---|---|
| 1 | 160691 | 160789 | 50 | 64 | 166 | 19 | ‘-‘ 15 |
| 2 | 160691 | 160789 | 51 | 65 | 60 | 19 | ‘-‘ 15 |
| 3 | 160691 | 160789 | 69 | 92 | 1 | 19 | ‘-‘ 24 |
| 4 | 160691 | 160789 | 55 | 72 | 33 | 19 | ‘-‘ 18 |
| 5 | 160691 | 160789 | 55 | 69 | 6 | 19 | ‘-‘ 15 |
| 6 | 160691 | 160789 | 54 | 71 | 7 | 19 | ‘-‘ 18 |
| 7 | 160691 | 160789 | 52 | 66 | 7 | 19 | ‘-‘ 15 |
| 8 | 160691 | 160789 | 53 | 67 | 2 | 19 | ‘-‘ 15 |
| 9 | 160691 | 160789 | 53 | 70 | 2 | 19 | ‘-‘ 18 |
| 10 | 160691 | 160789 | 52 | 69 | 3 | 19 | ‘-‘ 18 |
| 11 | 160691 | 160789 | 54 | 62 | 2 | 19 | ‘-‘ 9 |
| 12 | 160691 | 160789 | 54 | 68 | 1 | 19 | ‘-‘ 15 |
| 13 | 160691 | 160789 | 60 | 68 | 1 | 19 | ‘-‘ 9 |
| 14 | 160691 | 160789 | 68 | 91 | 2 | 19 | ‘-‘ 24 |
| 15 | 167262 | 167447 | 25 | 26 | 2 | 20 | ‘-‘ 2 |
| 16 | 160691 | 160789 | 51 | 56 | 1 | 19 | ‘-‘ 6 |
| 17 | 159890 | 160012 | 96 | 97 | 1 | 18 | ‘-‘ 2 |
| 18 | 160691 | 160789 | 53 | 62 | 1 | 19 | ‘-‘ 10 |
| 19 | 160691 | 160789 | 69 | 70 | 1 | 19 | ‘-‘ 2 |
| 20 | 160691 | 160789 | 50 | 51 | 2 | 19 | ‘-‘ 2 |
| 21 | 160691 | 160789 | 55 | 66 | 3 | 19 | ‘-‘ 12 |
| 22 | 160691 | 160789 | 44 | 51 | 1 | 19 | ‘-‘ 8 |
Input parameters for the EGFR gene exons 18, 19 and 20 used in pre-processing algorithm
2.2. General limitations of classifications methods
In general, the identification of an unknown sample is limited by the sample size and sample’s distribution homogeneity on which classifier is based on. The classification (mapping) which is based on a quantity-dimension class maps n data from an input space into m class of out space of defined classifier (Artificial Neural Networks, or Self Organizing Maps or some another clustering methods) where n (all combinations of mutated EGFR exons) in our case is much larger then m. Exons 18, 19, and 20 have a different number of nucleotides: 123, 99, and 186 respectively.
Certain exons may have either microdeletion mutations or no microdeletions, representing two states of an exon. The total number of combinations in an exon is dependent on their length, and for exons 18, 19, 20, and 21 can be represented as: n18 = 2123, n19 = 299, and n20 = 2186. Statistically known mutations for exons 18, 19, 20 are m18 =1, m19 =20, and m20=1. The number of statistically known mutations of exons mi (i= 18, 19, 20) are small compared to the total number of combinations of mutated exons ni (i = 18, 19, 20). The classifiers trained (learned) by input set (CTIS):
Training set is formed with the number of unique records (number of mutations + 1 exon without mutations) for each exon: 18, 19, and 20, respectively (1+1), (20+1), and (1+1) records [26].
These limitations motivated us to define our own goal, i.e., to develop a fully automated computer model that generates highly reliable identification data (of 18, 19, and 20 exons) for use in clinical applications. For such a model to be successful, it must implement the best possible reliable methods. Hence, the goal of development of such a model was to change strategy from approximate prediction based on classifier to exact identification that will operate using a “predictive” microdeletion mutations’ database for 18, 19 and 20 exon. Our model would then be able to identify the exact structure of mutated EGFR gene of clinically (statistically) known mutated exons and clinically not known (non-statistically known) mutated exons given in data sets of equation (3) and so achieve maximum values of sensitivity and specificity.
2.3. Approach based on knowledge base identifier of “predictive” deletion mutations
Due to the poor results (training, validation, and test errors) of approximate classifier in the [26], we developed the model based on generators of “predictive” combinations of all microdeletion mutations on exons 18, 19 and 20. This approach opens the following question: how to generate all mutations’ combinations on an exon in two states (with and without microdeletions) and to achieve a complete knowledge base of “predictive” mutated exons within the limits of generator time and computational resources.
To answer the above question we shifted our focus to the processing of genes based on exact identification for any sample from the set of all combinations of mutated exons.
We consider the microdeletions in mutated exons. Therefore, in a new mutated exon there are two possible states for every nucleotide. These states depend on nucleotide existence in mutated exon resulting in 2n possible mutated exons, including the original one, where n represents number of nucleotides in an exon. Exons 18, 19, and 20, have 123, 99, and 186 nucleotides respectively. The number of possible mutated exons is:
The development of a microdeletion mutation generator covering all combinations of distributed nucleotide mutations related to the positions in exons and the number of mutated nucleotides would be a very long process and not useful in real time.
In case of a “little” exon (exon 19), we have 99 nucleotides where a microdeletion mutation can occur (nucleotide dropping). The number of possible mutations is 299 ≈ 6.338253 ∙ 1029. If we assume a computer can generate 1 trillion (1012) microdeletion mutations per second (one of the fastest supercomputers in the world can do around 33.86 petaflops per second ≈ 33,860 trillion operations per second [29]), we would need around total time for the generation of n mutated exon 19 (TTGnexon19)
If we consider the Estimated Age of the Universe (EAU) is
We can see that a computer with 1 trillion mutations per second is not useful in real time.
2.4. Knowledge based generator of microdeletion mutations
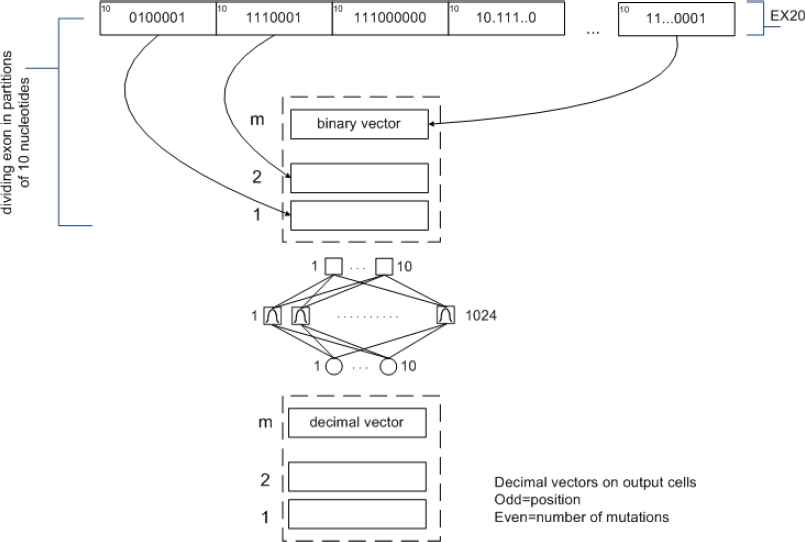
We developed another approach for solving the current problem based on mutations’ combinations related to parts of exon. Instead of working with the whole exon, we partitioned the exons and searched for mutations on those partitions. After identification process of microdeletion on exon parts, we integrate that partial combination results into whole exon. Specifically, as exon of n nucleotides can have 2n different possible mutations (microdeletions), we worked with groups of 10 nucleotides. With 10 nucleotides, there are 210=1024 different mutations, which are easier to recognize.
If exon having length n nucleotides and s parts, each part comprising 10 nucleotides, than the complete number of combinations is made as shown in Fig. 2.
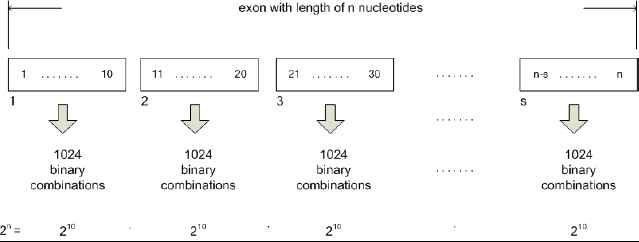
Combination of input space vectors for the exon having length multiple of 10 nucleotides
In case exon is having length m nucleotides, comprises of k-1 parts with 10 nucleotides each, and if kth part has l nucleotides (l < 10), this last part would be extended with x=10-l nucleotides to enable identification algorithm in the parsing phase to take into account last cycle of encoding with the RBNN. During the integration phase, identification algorithm would not take into account these additional 10-l nucleotides. Formation of a whole set of combinations for this case is presented in Fig. 3.
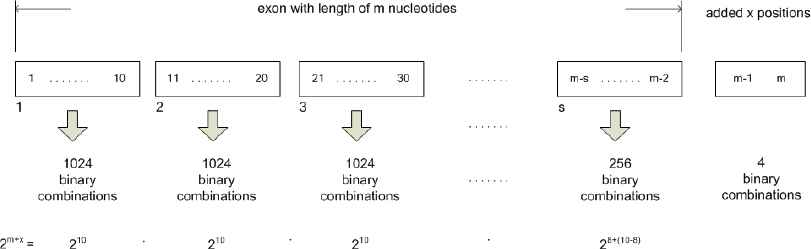
Combination of input space vectors for the exon having length not being a multiple of 10
These systems work best when many training vectors are available. In our research, the radial basis networks consist of two layers: a hidden radial basis layer of S1=1024 neurons and an output linear layer of S2=10 neurons with bias b1 vector and bias b2 vector, which can lead to the first and second layer respectively (Fig 4.)
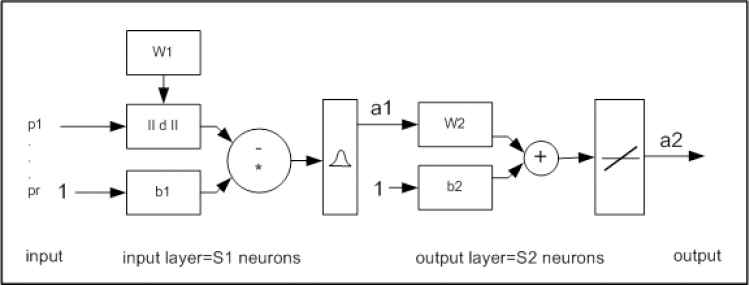
RBNN structure with input (RADBAS) layer and output (linear) layer
Further, the RBNN with 1024 cells (in the first layer) was designed with each cell representing a unique mutation structure (class) on length of 10 nucleotides. The RBNN is an artificial neural network that uses radial basis functions as transfer functions.
The knowledge base consisted of 1024 classes of mutations, which were used repeatedly n/10 times puta (n is number of nucleotides in an exon) for recognition of mutations in one complete exon.
The RBNNs can require more neurons than the standard feed forward backpropagation networks, but they can be designed in a fraction of the time it takes to train the standard feed forward networks.
In this research radial basis network was designed using the newrbe function. This function can produce a network with very small error for each component of any vector in the output matrix, thus all output values are deterministic and predictable. The following instructions are related to a network with exact number of neurons (where number of input vectors from input matrix is defined in advance), and to a network with fewer number of neurons (network is trained with a fewer number of neurons compared to the number of input vectors of the input matrix).
The functions newrbe i newrb take matrices of input vectors P and target vectors T, and a spread constant spread for the radial basis layer, and returns a network with weights and bases such that the outputs are exactly T when the inputs are P. This function creates as many RADBAS neurons as there are input vectors in P, and sets the first-layer weights to P’. Thus, there is a layer of RADBAS neurons in which each neuron acts as a detector for a different input vector. If there are Q input vectors, then there will be Q neurons. The next equations are used to simulate work of RBNN for test input p in exact number of neurons (12) and in fewer neurons (13).
The following instructions are used to evaluate validity of network, and we expect that difference between the any coordinate of the output test vector (in output test matrix) and the any appropriate coordinate of the target vector (in the target matrix) should not exceed 0.5 in absolute value.
It is important that precision of the value obtained by coordinates of the vectors in output matrix is sufficient to determine exact number approximated by the output value. Here is an example:
- •
Input vector from input matrix: [1 0 1 1 1 1 0 0 1 1]
- •
Output vector from output target matrix: [2 1 6 2 0 0 0 0 0 0])
- •
Output vector from output matrix obtained in the test (simulation) process [2.397 0.5111 5.999 2.494 0.00003 0.0124 0.468 0.245 0.354 0.2125]
Error for each coordinate between output target matrix and output test matrix needs to be smaller than 0.5 in order to obtain correct output when rounded.
3. Model of the system for EGFR microdeletion mutations analysis
Development and implementation of the system model (Fig. 5) were done in the MATLAB environment using interactive graphical user interface (GUI) for setup, processing in training mode and exploitation mode. In the beginning of the processing, following options are selected:
- –
Mode: Training Processing Mode-TPM, Exploitation Processing Mode-EPM,
- –
Data source: NCBI database, clinical database, local database of technologically extracted and processed EGFR genes, and
- –
Exon: defining exon in focus for mutated nucleotides identification.
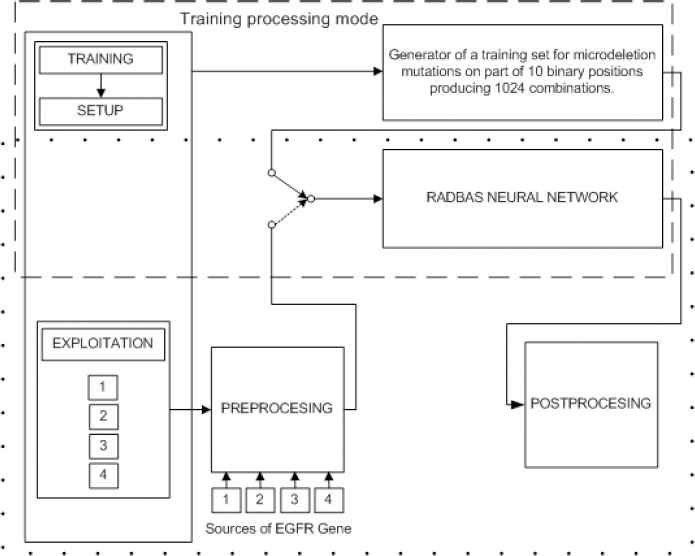
Processing modes of system model
3.1. Design and training of RBNN
In this mode, RBNN is used for training/learning with the training sets produced by combinatorial generator of microdeletion mutations. The “predictive” data set generation strategy builds large data sets for training, as there are many combinations of mutations for each exon making the training of the ANN challenging from hardware and design perspectives.
3.1.1. Generation of training set
As we said in part 2.4 instead of working with whole exon, we solved this problem by partitioning exons in parts and looking for mutations on those parts. However, the “predictive” mutation database for exact identification had to be prepared first. As an exon of n nucleotides can have 2n different possible mutations we worked with groups of 10 nucleotides
If we look for mutation in the shortest exon, in our case, we would have to examine positions of 99 nucleotides. Implementation of a model to identify missing nucleotide would require 299 different combinations, with high requirements for resources and time. This was a reason to examine not whole exon, but only the parts of exon, with size limited to 10 nucleotides, leaving the last part with size less or equal 10. This approach leads to examination of 210 = 1024 combinations, i.e. 1024 binary input vectors. For each binary input vector there is a corresponding decimal output (class) representing the individual combination of mutations in the 10-nucleotide sequence of the exon.
The generation of a training set (input_matrix and output_matrix) for the RBNN in exact training mode (target error = 0, neuron number in hidden layer is 1024) was developed by the next generator functions using data base with binary notations of EGFR exons. Pseudo code for input matrix generate function is:
Algorithm 1:
input_matrix = generate_01_matrix(10)
generate_01_matrix (input_size)
generate input_matrix of size (input_size x (2^input_size))
for every column
replace column with binary representation of column index
return input_ matrix
Output target matrix is generated using function generate_01_matrix_RADBAS_output. Output vectors contain information on deletions, non-zero odd elements indicate position of a nucleotide where deletion starts, and subsequent even position indicates number of deletions. If input column is:
Pseudo code for output matrix generate function is:
Algorithm 2:
output_matrix =
generate_01_matrix_RADBAS_output(input_matrix);
generate_01_matrix_RADBAS_output( input_matrix )
**generate output matrix with proper dimensions**
for every column of input matrix
replace matching column of output matrix with proper
result
return output_matrix
Vectors in this matrix are reflecting deletions distributions. The following pseudo code is presenting how output vectors are generated:
Algorithm 3:
proper_result( input_vector )
if input vector dimension is even then
generate output_vector with dimension equal to that dimesion
else
generate output_vector with dimension greater for one of input vector
start from begining of input vector
repeat until end of input vector
find first next zero value
replace first next value in output vector with that value
count number of consecutive zeros in input vector from faunded value
replace first next value in output vector with that number
end of repeat
Generating functions produce training set containing input and output matrix. Input matrix contains 1024 binary input vectors; output matrix contains respective 1024 decimal vectors with length 10, each providing unique information on position and region of mutations
3.1.2. Different training modes for RBNNs and result analysis
Mode exact_training_mode is not a typical training of neurons, but during a single iteration (one epoch) with function newrbe all neurons are created with weights determined by values of respective input vector. Syntax of this command is:
Mapping of one part of input matrix into a part of output matrix in exact_training_mode is shown in Fig. 6. Ten input vectors consist of 10 binary elements (0 = mutation, 1 = no mutation) (Fig 6.a) and 10 output vectors with decimal identification of positions/number of mutations (Fig. 6 b). Odd number on RBNN output vector indicates the beginning of a mutation sequence, and even number on RBNN same vector indicates the number of mutations from that position.
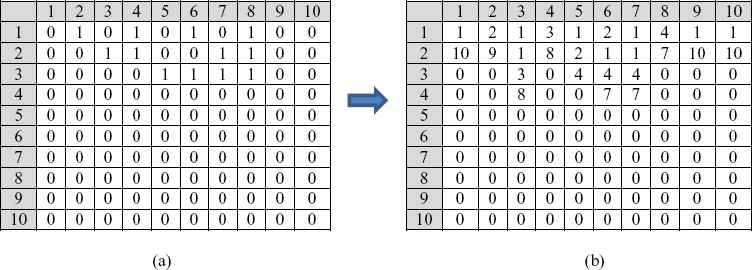
(a) 10 binary input vectors from 1024 binary combinations of mutations, (b) 10 corresponding output vectors from 1024 decimal combinations of classes related to identification of mutations.
For example, first input vector has 10 mutations and first output vector has on odd output number value 1 and on even output number value 10, what means that we on first part of one exon have ten mutations starting from first position. This information will be very important in the process of calculating distribution of mutations on complete exon (relative positions) and complete gene (absolute positions).
Training process resulted in a network shown in Fig. 7
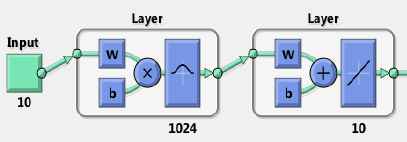
RBNN obtained by newrbe command in exact_training_mode
Because the RBNN property is 1 input (combination of ten binary numbers) to 1 output (combination of 10 decimal numbers) mapping learning mechanism forms 1024 unique RADBAS neurons (each for one exact class) and an outer layer with 10 linear neurons generating corresponding outputs. The result of the above process was a knowledge-based mutations RBNN in function of binary to decimal encoder named RADBAS_exact. Parameters of obtained network are W1_result, b1_result, W2_result and b2_result. This RBNN has objective to identify mutated exons on one exon. In order to implement the network in Simulink model function genism (RADBAS_exact) was used. Network simulation generated test output matrix:
Now it is possible to evaluate generated network comparing outpt_matrix and RADBAS_exact_output. Evaluation is not done by calculating total error, but we look for the maximal error in co-ordinates of output vectors, obtained as maximal absolute difference between elements in two matrices:
Value of the calculated error is:
We can conclude that the RBNN output is equal to desired output with precision of 12 decimal places. Flow diagram of design and training of knowledge-based mutations RBNN (RADBAS_exact) is shown in Fig. 8.

Flow diagram of design and training of the knowledge-based mutations RBNN
It would be tedious to generate RADBAS networks with different parameters: number of epochs, number of neurons, and maximal absolute error for each coordinate for each test output vector, and with respect to target matrix vector, the RADBAS network has different structure.
This was a reason to design combinatorial generator of RADBAS network parameters, including instructions (10), (11), (12), (13), (14) and (15). Pseudo code of the generator:
Algorithm 4:
function combinatorial parameters (input_matrix, output_matrix, mse, spread, max_rb_neurons, step)
generate output as matrix with (dimension of mse * dimension of spread) rows
for every mse
for every spread
train RADBAS ANN with specific mse and spread
replace next row of output with [current_mse, current_spread, number_of_RADBAS_neurons, maximum_of_absolute_error_between_output_matri x_and_simulated_results]
return output;
Selected resulting performance diagrams are presented in Fig. 9 and Fig. 10.
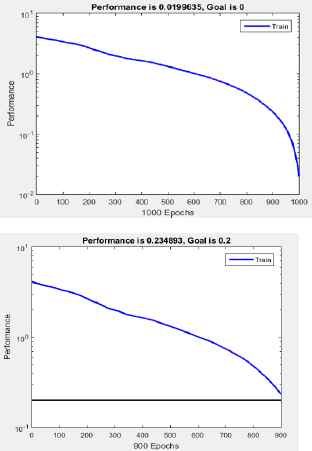
Performance of RBNN generated with: a) newrb(), goal error 0, and structure with less than 1024 neurons, b) newrb(), goal error 0.2, spread constant equal 0.3, and structure with less than 1024 neurons.
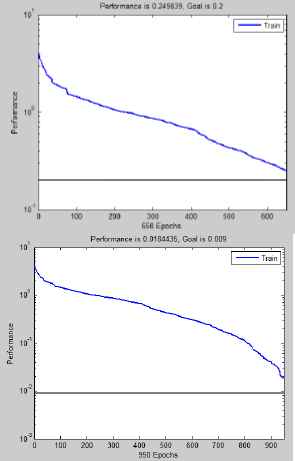
Training performance of RBNN generated with: a) newrb(), goal error 0.2, spread constant equal 3, and structure with less than 1024 neurons, b) newrb(), goal error 0.009, spread constant equal 3, and structure with less than 1024 neurons.
The obtained network has following parameters: goal error = 0.009, and spread constant=3, and close to 1000 RADBAS neurons, but errors for co-ordinates are not smaller than 0.5. We can conclude that even if we succeed in obtaining a network with a number of neurons less than 1024, we do not achieve significant reduction in computational resources devoted to the network. In the mode exact_training_mode, in one iteration we obtain a network with 1024 neurons, and absolute error for co-ordinates of all output vectors is smaller than 0.5 (3.0871e–13).
In the mode fewer_trainig_mode, with a goal error 0, network is again constructed with 1024 neurons, absolute errors for co-ordinates of all output vectors are again smaller than 0.5 (2.6449e–13). Knowing that exact mode training is performed in one iteration with determined outputs containing distribution of deleted nucelotides, in this research we decided to employ RBNN created in the exact_training_mode. Table 2. provide results from several experiments.
| Method and parameters | Name | Number of epochs | Number of RADBAS neurons | MSE | Maximal absolute error for coordinates of output test vector compared against the target output vector |
|---|---|---|---|---|---|
| exact_training_ mode | RADBAS_exact_1024 | 1 | 1024 | - | 3.0871e–13 <0,5 |
| fewer_training_mode goal error =0, spread=0,3 | RADBAS_fewer_0_03 | 1025 | 1024 | 0.019635 | 2.6449e–13 <0,5 |
| fewer_training_mode goal error =0.2 spread=0,3 | RADBAS_fewer_02_03 | 915 | 914 | 0.234893 | 6.0789>0,5 |
| fewer_training_mode goal error =0,2 spread=3 | RADBAS_fewer_02_3_0 | 697 | 650 | 0.249839 | 3.9394>0,5 |
| fewer_training_mode goal error =0,009 spread=3 | RADBAS_fewer_0009_3_0 | 968 | 950 | 0.184435 | 0.7729> 0,5 |
Performance results of RADBAS neural networks for different parameters and structures
3.2. Exploitation of the system
The second mode is related to the exploitation of the system, which can perform off-line or on-line processing of EGFR genes. EGFR genes for off-line processing can come from different sources. Real time processing of EGFR genes uses EGFR genes in a text format obtained from diagnostic process, from biopsy to DNA extraction. Both processing modes involve data preprocessing and identification of distributed mutations. Detailed operational procedure for the system model in exploitation is shown in Fig. 11.
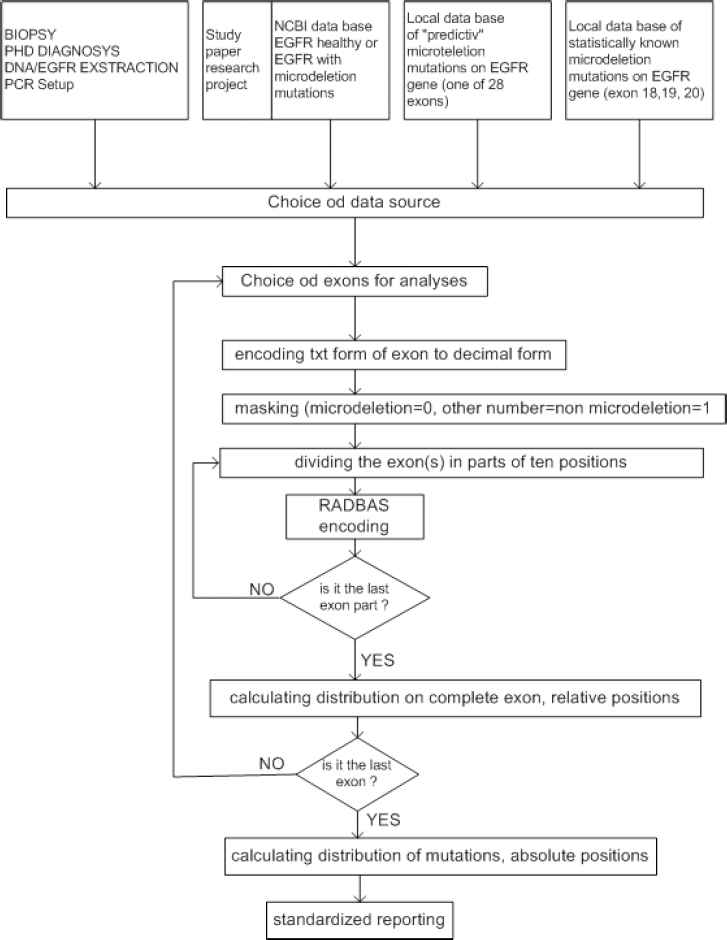
Detailed operational procedure for the system model in exploitation
3.2.1. Acquisition and setup
In off-line mode the system can process EGFR genes from the following sources: (1) NCBI data base, (2) clinical data base with the statistically known mutated exons, and (3) local data base with the predictive data base of mutated exons. In real-time mode EGFR gene is obtained from (4) technology procedure: biopsy, diagnostics, DNA/EGFR extraction, DNA/EGFR quantification, PCR procedure, resulting in a text notification of nucleotides in EGFR sequence (Fig. 12).
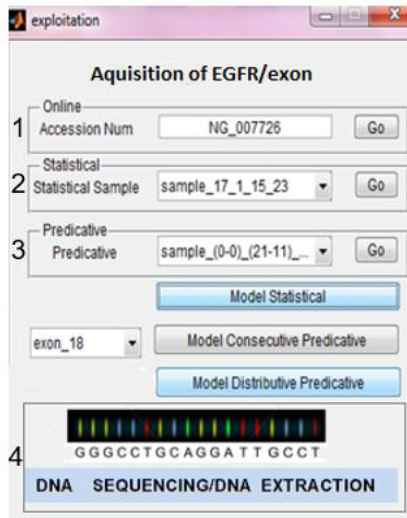
Different sources for mutated EGFR genes
3.2.2. Preprocessing
Both modes include preprocessing of data (extraction, encoding, and masking) (Fig. 13), identification of distributed mutations (RBNN encoding, counting of exon mutations distribution and counting of EGFR gene mutation distribution), and standard reporting.
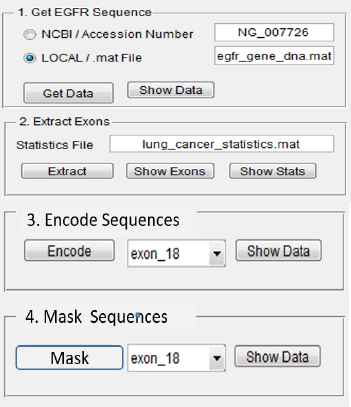
Graphical user interface for data preprocessing
EGFR gene DNA sequence is obtained from one of the sources, and exons of interest are extracted. For this system the genes 18, 19 and 20 are extracted. Nucleotides a, c, g, and t are encoded into numeric values as numeric format is easier to process by computer algorithms. Table 3 shows encoding table rules for each nucleotide sequence. The dash symbol (-) represents a microdeletion mutation of the DNA sequence.
| Patient | Gene | Exon | Relative position (within exon) and number of mutations | Absolute position (within gene) and number of mutations | Clinically known mutation (statistics) | First occurrence of identified mutation |
|---|---|---|---|---|---|---|
| 1 | EGFR | 19 | 54/18 | 160745/18 | Yes | No |
| 2 | EGFR | 19 | 1/10 | 160691/10 | No | Yes |
| 3 | EGFR | 18 | 96/2 | 159986/2 | Yes | No |
| 4 | EGFR | 20 | 25/2 | 167287/2 | Yes | No |
| 5 | EGFR | 19 | 12/9, 21/1, 23/8 | 160703/9, 160712/1, 160714/8 | No | Yes |
Standard report of gene mutations related to distribution and occurrence
3.2.3. Identification of microdeletions
Identification of microdeletions is conducted with a function: complete_exploatation_for_one_exon, which involves the sequence of functions: input vector masking, (mask input vector) within the framework of an exon, parsing masked vectors (parse masked vector) within the framework of the masked exon, and preparation of deletion distribution for a complete exon (reassemble result). Pseudo code for the identification process:
Algorithm 5:
complete_exploatation_for_one_exon(input_exon )
get size of input vector as vec_size
mask input vector as binary vector, 1 for O, and 0 for missing
parse masked vector as matrix of dimension 10 x ⌈vec_size / 10⌉
get simulation of parsed matrix with RADBAStrain
reassemble result of simulation
prepare output of reassembled simulation
Identification of microdeletions is illustrated using exon 19. Exon 19 has 9 mutations on positions 54–62 (Table 1). After reading, the exon 19 non-mutated nucleotides are masked with 1, and mutated positions with 0. Masking input exon 19 is presented with the following pseudocode:
Algorithm 6:
mask_input_19 (input_vec)
set output_vec equal to input_vec
in output_vec, change all non zero values to 1
return output_vec
Parsing of input masked exon results in producing m x 10 vectors (Fig. 14) and is presented with the pseudocode:
Algorithm 7:
exon_parsing_19( vector, parsing_size )
add ones to end of vector to have a size divisible by parsing_size
generate output matrix of dimension 10 x ⌈vec_size / 10 ⌉
for every 10 size subvector of input vector
replace next coumn of output matrix with current subvector
return output matrix
Example of parsing input masked exon 19
The conversion of masked exon to matrix format is given with
For example, exon 19’s length is 99. We will use the generated RADBAS N times, with N as the result of an expression:
Since exon 19 length is not multiple of 10 nucleotides, we would add one nucleotide to the exon resulting in a number of nucleotides divisible by 10, and after counting microdeletions on complete exon we would remove that nucleotide from the exon’s original length. In other words, if exon length is n, than we have: n = 10 ∙ k + l where k is integer quotient of dividing n by 10, and 0< l< 10 is the reminder of division.
After identification of mutated nucleotides, output vector of RADBAStrain neural network is integrated and presentation of deletions distribution for complete exon is prepared, as described with the following pseudocode:
Algorithm 8:
result_reassembling( input_matrix, parsing_size, vector_size ))
generate vector with vector_size from adding columns from input matrix
adding parsing_size*column index to all non zero numbers on odd position
of column element
repeat
if vector has zero elements between non-zero elements, move non-zero
elements to lower positions
for every non-zero element on odd position (say k)
if next element on odd position (k + 2) is non-zero and equal to
sum of preceding two elements, then element on position (k + 1)
increment with value of element on position (k + 3) and then set
to 0 elements on positions (k + 2) and (k + 3)
until no change has made
Positions of mutated nucleotides are given relative to the beginning of the EGFR gene. Fig. 15 shows transformation from positions within the exon parts to positions within the exon 19 for a patient.
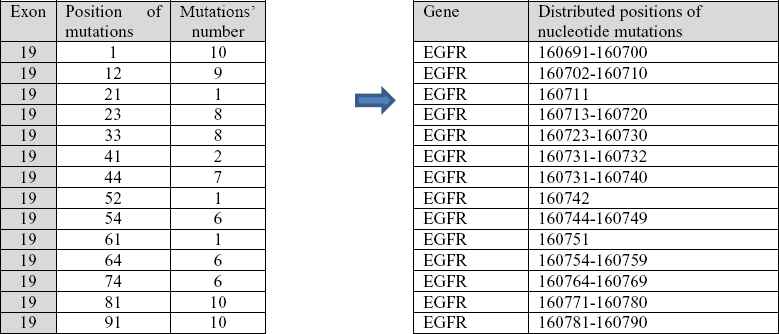
Identified mutations in distributed regions of exon 19.
After transforming positions and number of mutations within exons, algorithm continues with transforming positions of mutated nucleotides within the EGFR gene. Algorithm creates two special arrays with a length equal to number of exons in the gene. The first array contains positions of the first nucleotide in each exon - gene_exon_first_nucleotide , and the second array contains positions of the last nucleotide in each exon - gene_exon_last_nucleotide. The arrays enable enlisting of all mutations (deleted nucleotides) for a whole EGFR gene. Pseudo code to calculate and display mutated nucleotides within the EGFR gene is described with the following pseudo code:
Algorithm 9:
gene_mutation_summary (
array_of_exon_reassembling_results )
**generate result_matrix as empty matrix**
for every matrix in array_of_exon_reassembling_results
set index to be index of element in array
create temp_matrix equal to matrix from array on position index
modify temp_matrix’s first column by adding
(gene_exon_first_nucleotide[index] - 1) to every element in first
Column vertically append temp_matrix to result_matrix
repeat
if vector has zero elements between non-zero elements, move non-zero
elements to lower positions
for every non-zero element on odd position (say k)
if next element on odd position (k + 2) is non-zero and equal to
sum of preceding two elements, then element on position (k + 1)
increment with value of element on position (k + 3) and then set
to 0 elements on positions (k + 2) and (k + 3)
until no change has made
Result of running the algorithm based on the above pseudocode; on exon 19 are absolute positions of mutated nucleotides (Fig. 16).
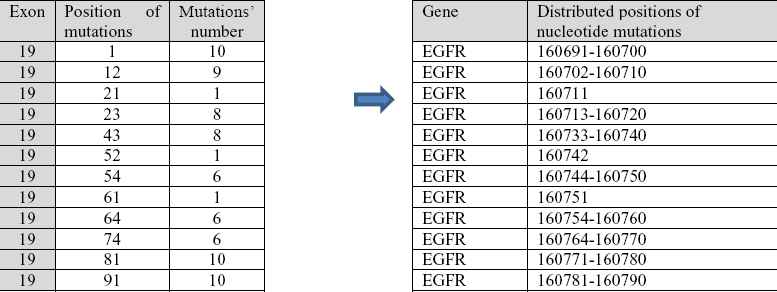
Identified mutations in distributed regions of complete gene EGFR
The last step of identification is to generate a standard report containing data as shown in Table 2. for patients 1 and 5.
Example for the patient 1:
On the exon 19 of the EGFR statistically known mutations were identified at absolute location within the gene: 160741-160756. For the mutations there is clinically established therapy based on gefitinib, i.e. erlotinib [34].
Example for the patient 5:
On the exon 19 of the EGFR statistically unknown mutations were identified at absolute location within the gene: 160703-160712, 160712-160713 i 160714-160722. New therapy should be established for these mutations.
Described code could be used for any gene, and parameters for the program are exon number, absolute address of the beginning and the end of each exon. Depending whether position of data in the array gene_exon_first_nucleotide and in the array gene_exon_last_nucleotide is given in relation to gene or genome, exploitation algorithm will in the last step provide solution containing mutations index within the gene, or genome.
4. Discussion
In this paper has described solutions based on RBNN identifier using training set generated with the combinatorial microdeletion mutations generator with the purpose to compensate unreliability of classification results in available literature, and generally compensate for computer resources and real time computing.
Our approach introduces the exact identification of microdeletion mutations. The mutations identified includes statistically known mutations, consecutive mutations in one region, and randomly distributed mutations in several regions using the knowledge base generator implemented with RBNNs and MATLAB s-functions.
The benefit of this approach is exact identification of positions and distributions of mutations, what was proved on complete sets of patient classes and using binary mutation combinations generator developed within this research.
This research raises the following questions: Why did we use computing system model based on RBNNs and s-functions, rather than classical procedures written in a programming language?
The answer consists of the following:
- •
First, we wanted to show the flexibility and robustness of the application of RBNNs in two phases. In the design phase, RBNN is used as a generator of predictive mutations’ partitions of 10 nucleotides of a particular exon. In the exploitation phase, the same network is used for the identification of mutated genes in a particular exon of EGFR gene.
- •
This is a generic type model and can be applied (with minor parametric modification) to any exon in any gene. That is, it can be applied to all exons in a single gene, and all genes in one genome, but only when you want to identify the microdeletion mutation type. Therefore, we want to achieve tractability, robustness and low solution cost.
- •
The model is open source, which means that we can at any preprocessing step (extraction, encoding, masking) and at any processing step (parsing of exons, identification of deletions, calculating of deletions relative positions and counting of deletions absolute positions on complete EGFR gene) follow graphically parallel results in all mutated exons and make appropriate conclusions.
- •
Today, for the analysis of EGFR gene mutations, we need senior pathologists, which are rare, so reliable identification of mutated exons is not always available. Therefore, this approach offers analysis and decision support to clinicians and assists in education of young pathologists in exact identification of distributed mutations looking at the same time in all wanted exons.
- •
This approach present exact deterministic identification of each nucleotide deletion.
5. Conclusion and future research
The computing system model for EGFR microdeletion mutations analysis is only a part of a larger research project “Intelligent Decision Support System for Lung Cancer Diagnosis, Survival Prognosis, Treatment Monitoring, and Biomarkers Discovery Using Fusion of Artificial Intelligence Methods and Gene Microarray Technologies in the Area of Personalized Medicine [30].
The overall goal of this study is to show that the combination of patients’ clinical and genetic data can significantly improve decision support systems’ predictive performance for lung cancer.
Bearing in mind our goal to develop solutions applicable to personalized medicine, our conclusion is that is necessary to determine the structure of mutated EGFR genes for each lung cancer patient: mutated exon(s), the position of the mutation(s) and the number of mutations.
When knowing the exact structure of mutations in exons, pharmacists, molecular biologists, chemists, clinicians, therapists, and bioinformaticians (united in one team) can determine the appropriate medication and therapy. As each lung cancer patient can have several mutated genes for one kind of lung cancer determined as biomarker, our next step is to make an extension of this approach to all these genes.
In this way, it can be easily analyzed at the same time, allowing mutations’ detection, if there are more biomarker-genes causally acting on a specific lung cancer type [31]. For this, we would develop a new ensemble classifier using RBNN [32] in order to increase higher detection rate of microdeletions on more than one gene.
This model is intended specifically for the identification of microdeletions’ mutations, and it would be interesting to expand this principle of RBNN identifier to other types of mutation regions of a gene, like converting the nucleotides between them, deep intronic and promoter mutations [33], and integrate it in one clinical decision support system [34]
Contributions
ZA conceived and designed the research. ZA and DB evaluated appropriate artificial methods employed in this research. VL devised mathematical model for combinatorial binary generator and RADBAS Neural Network encoders. ZA and DB drafted earlier versions of the manuscript. AS performed acquisition and analysis of material from the field of molecular biology and oncology for the lung cancer. All authors read, reviewed and approved the final manuscript. All of the authors agreed on the content of the paper and declared no conflict of interest.
Acknowledgments
We thank BiH Federal Ministry of Education and Science for the financial support in 2012/2013 for the research project “Computer Aided Lung Cancer Classification of Mutated EGFR Exons Using Artificial Intelligence Methods” conducted at the Faculty of Electrical Engineering University of Sarajevo, and for the financial support in 2014/2015 for the research project “Computer Based Modeling In Bioinformatics For Gene Based Cancer Classification Focused On Reliability And Machine Learning” in international collaboration between University of Sarajevo-Bosnia and Herzegovina and University of Ljubljana-Slovenia.
References
Cite this article
TY - JOUR AU - Zikrija Avdagic AU - Vedad Letic AU - Dusanka Boskovic AU - Aida Saracevic PY - 2018 DA - 2018/07/26 TI - EGFR Microdeletion Mutations Analysis System Model Using Parameters Combinations Generator for Design of RADBAS Neural Network Knowledge Based Identifier JO - International Journal of Computational Intelligence Systems SP - 1248 EP - 1267 VL - 11 IS - 1 SN - 1875-6883 UR - https://doi.org/10.2991/ijcis.11.1.93 DO - 10.2991/ijcis.11.1.93 ID - Avdagic2018 ER -