
Does Treatment for Obstructive Sleep Apnoea Improve Arterial Stiffness? Evidence from Randomized Clinical Trials on Carotid-femoral Pulse Wave Velocity
 , C. Fava1, M. Pengo3, P. Chowienczyk2, J.K. Cruickshank4
, C. Fava1, M. Pengo3, P. Chowienczyk2, J.K. Cruickshank4- DOI
- 10.2991/artres.k.201102.004How to use a DOI?
- Keywords
- Obstructive sleep apnoea; arterial stiffness; cardiovascular diseases; c-PAP; risk factors; vascular function; pulse wave velocity
- Abstract
Obstructive Sleep Apnoea (OSA) is a breathing disorder characterized by narrowing of the upper airway that impairs normal ventilation during sleep. OSA is a highly prevalent condition which is associated with several Cardiovascular (CV) risk factors and CV diseases. Despite this clear association, Randomized Controlled Trials (RCTs) have provided equivocal data that treatment of sleep apnoea can improve CV outcomes regardless of its ability to reduce blood pressure. Here, we critically review the evidence that supports role of OSA as a risk factor for increased arterial stiffness which represents an early manifestation of vascular damage often preceding major CV events. Additionally, we examined evidence from interventional RCTs to assess if treatment of OSA by continuous positive airway pressure can affect arterial stiffness measured as carotid-femoral pulse wave velocity. Overall, a large body of evidence supports the role of OSA as a risk factor for increased arterial stiffness and several pathophysiological mechanisms, including activation of the autonomic nervous system, may help to explain the link between breathing disorders and vascular alterations (here mainly examined as functional properties). Whether the causal relationship between OSA and vascular damage exists or is mostly explained by confounders and whether OSA treatment can improve vascular stiffening is still debated.
- Copyright
- © 2020 Association for Research into Arterial Structure and Physiology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. OBSTRUCTIVE SLEEP APNOEA: AN OVERVIEW AND ITS ASSOCIATION WITH CARDIOVASCULAR RISK FACTORS
Obstructive Sleep Apnoea (OSA) is a breathing disorder characterized by narrowing of the upper airway that impairs normal ventilation during sleep [1]. Patients may present with daytime symptoms (such as excessive daytime sleepiness, poor concentration and fatigue secondary to sleep disturbance); nocturnal symptoms (such as snoring, frequent awakenings, choking sensations) or can be asymptomatic. The prevalence of OSA varies significantly based on the cohort being studied [2], and in western countries is estimated between 2 and 4% of the general population [3]. The prevalence of OSA is also projected to increase largely as a result of the obesity epidemic in both developed and developing countries [4] since an estimated 50% of obese individuals with metabolic syndrome and/or diabetes [5] are affected to some degree [6,7]. Furthermore, the increase of childhood obesity leads to rising prevalence of sleep-disordered breathing even in the young where an association with early sign of vascular damage at carotid level has also been detected [8]. The prevalence of OSA in the general population is likely to be underestimated since symptoms of OSA are often nonspecific and polysomnography, the gold-standard test for its diagnosis, has limited availability. With polysomnography, the OSA syndrome can be diagnosed when the Apnoea–Hypopnoea Index (AHI) is >5 events/h associated with the typical symptoms or ≥15 events/h without symptoms [2].
Several treatments for OSA have been proposed over the years including oral devices in mild forms [9] and Continuous Positive Airway Pressure (c-PAP) in moderate/severe cases. Lifestyle modification aimed at weight reduction is always recommended alongside treatment, but a referral for bariatric intervention, such as intragastric balloons or surgery, may be needed to achieve significant weight loss and improve the severity of OSA in morbidly obese subjects [10].
Continuous positive airway pressure and other treatments are not only effective in reducing AHI and alleviating symptoms but also appear to ameliorate the Cardiovascular (CV) risk profile of the affected subjects. A large body of evidence shows that patients with OSA often present with increased inflammatory markers [11], endothelial dysfunction [12], raised arterial stiffness and Blood Pressure (BP) [13], probably sequentially, all contributing to a substantial excess in CV risk. Despite the well-recognized association between CV risk factors and OSA, whether a causal relationship exists or is explained by confounders [14] is still debated although more evidence seems to support its independent role both for CV risk factors [15] and CV morbidity and mortality [4].
In patients affected by OSA, the increased CV risk is mostly related to acute myocardial infarction and/or stroke, which are not fully accounted for by other conventional risk factors [16,17]. Longitudinal data have also confirmed a dose-response association between sleep-disordered breathing severity and Hypertension (HT), a relationship which is independent of well-known confounders [15]. This can then explain, at least in part, the excess of CV morbidity from heart failure, coronary artery disease [18] and stroke [17,19] in the population affected by OSA. Pathophysiologically, several mechanisms can support a causal relationship between OSA and CV risk factors, such as intermittent hypoxia [20], sympathetic activation and inflammation [4,21].
2. OBSTRUCTIVE SLEEP APNOEA AND ARTERIAL STIFFNESS
Vascular stiffness may be the key component in linking breathing disorders with CV morbidity [22] as observed in other respiratory conditions such as chronic obstructive airway disease [23]. Several mechanisms involved in OSA, including intermittent hypoxia, chemoreceptors, baroreflex stimulation and the activation of the autonomic nervous system may link this condition with the observed increased vascular stiffness [1] (Figure 1).
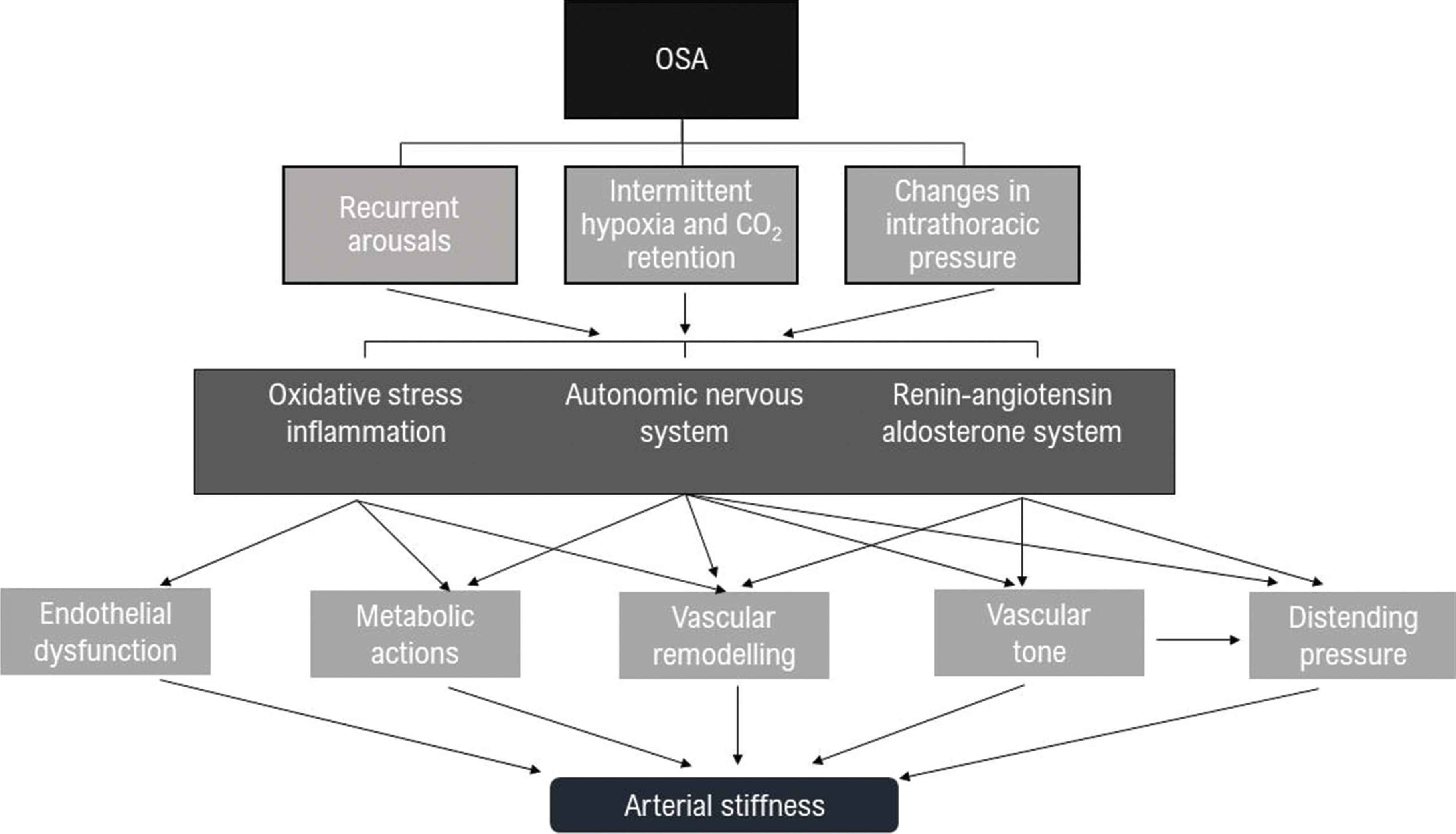
Simplified pathophysiological mechanisms that link Obstructive Sleep Apnoea (OSA) with increased arterial stiffness.
Reduced ventilation at night due to supine position can lead to intermittent hypoxia increasing oxidative stress and inflammation that is mediated by proinflammatory cytokines, adhesion molecules and procoagulant factors, all contributing to endothelial dysfunction, increased BP and early atherosclerosis [24]. At the same time, hypoxia and hypercapnia affects inspiratory effort and the magnitude of negative intrathoracic pressure against occluded airways which in turn increase left ventricular wall tension and myocardial oxygen demand [25]. Intermittent hypoxemia also activates the Renin–Angiotensin–Aldosterone System (RAAS), which is a key homeostatic pathway for BP regulation and is also implicated in cardiac [26] and vascular remodelling [27].
In this context, it is also important to recognize that a key component of the RAAS, Angiotensin II, exerts several actions on the sympathetic nervous system both centrally by increasing sympathetic outflow [28] and stimulatory effects on sympathetic ganglia and the adrenal medulla [29,30] and peripherally by facilitating neurotransmission [31,32]. Angiotensin II also interacts with baroreceptor reflexes to increase BP [33].
The autonomic nervous system is not only stimulated indirectly throughout the RAAS but also directly by recurrent arousals, alterations in intrathoracic pressure and intermittent hypoxia which can contribute to sympathetic overactivity [24] (Figure 1).
All the above-mentioned mechanisms promote endothelial dysfunction, metabolic alterations and vascular remodelling. Notably, they also influence BP regulation and since the distending pressure inside the blood vessel is a major determinant of arterial stiffness in vivo, they all contribute to the increased stiffness itself. However, apart from the BP-mediated effects, the autonomic nervous system can also play an independent role in the regulation of vascular stiffening as recently found in healthy individuals [34] and in subjects with HT [35]. At vascular level, the stress–strain relationship which determines arterial stiffness [36] is mostly attributed to the structural properties of the extracellular matrix rather than being influenced by the tone of vascular smooth muscle [37]. However, the latter has shown an independent action not only in muscular arteries but also in large elastic arteries (including the aorta) [35] included in direct assessment of arterial stiffness.
In this context, evidence of alteration in the autonomic nervous system have been documented in subjects affected by OSA including elevated circulating catecholamines [38] or increased muscle sympathetic nerve activity over 24 h compared to non-apnoeic controls [39]. Notably, some of the above-mentioned alterations in sympathetic activity can be ameliorated by the treatment in patients affected by OSA [40].
In summary, many of the determinants of arterial stiffness including increased BP, sympathetic activity and endothelial dysfunction can be directly affected by OSA. Cross sectional investigations suggest that OSA is independently associated with arterial stiffness, and patients in which OSA coexists with other well-established CV risk factors (such as HT) have the highest levels of arterial stiffness. However, this association does not prove causality and data from RCTs are conflicting [41].
3. DOES c-PAP TREATMENT AMELIORATE ARTERIAL STIFFNESS IN SUBJECT WITH OSA?
While the relationship between OSA and arterial stiffness was initially identified in small observational cohorts [42,43]; this was subsequently confirmed by a larger body of evidence showing a correlation between the severity of OSA and impaired vascular function [22]. However, strong evidence supporting the role of treatment, particularly c-PAP, on arterial stiffness is conflicting.
Examining data from observational studies, ultrasound measures of aortic elastic parameters, such as strain and distensibility, appeared improved by c-PAP treatment, although the findings did not take potential BP change into account [44]. Two other studies [45,46] showed use of c-PAP treatment to significantly decrease brachial-ankle pulse wave velocity with no effect on peripheral BP. Interestingly, one of these studies [45] also indicated that reduction in pulse wave velocity correlated with heart rate variability which is considered as a surrogate marker of autonomic nervous system activity. Despite some positive results, other researchers have failed to demonstrate an effect of OSA treatment on arterial stiffness [47] particularly after considering BP as a potential confounder [48].
A previously published metanalysis [22] performed in patients with OSA and HT suggested that c-PAP treatment can improve arterial stiffness. However, it included only one Randomized Controlled Trial (RCT) and three observational studies with limited data regarding BP change [22]. In their review, Philips et al. also reported that only a small number of studies examined the effects of treatment of OSA on arterial stiffness but the analysis had important limitations (including small sample sizes, lack of a control group and bias related to duration of interventions) [41].
The heterogeneity of these results can probably be related to the methodology used to assess arterial stiffness as well as differences in the study populations. To partially address these limitations, in this review, we have focused our analysis to only include interventional clinical trials in which arterial stiffness was measured as cfPWV. Arterial stiffness assessed by cfPWV is recognized as the non-invasive gold standard technique in vivo and is well established as an independent predictor of CV morbidity and mortality independently of BP [49,50]. Based on the meta-analysis of Ning et al. [51], four RCTs have investigated the effects of c-PAP treatment on arterial stiffness in adult population (Table 1) and a recent RCT in 2020 examined patient with resistant HT.
| Study | Population | Design | Duration of intervention | ΔPWV (m/s) | Device for PWV |
|---|---|---|---|---|---|
| Drager et al. [52] | 24 pt with severe OSA and no comorbidities or cardiovascular risk factor | Randomized, not-blinded | 4 months | −1.00 (1.19, −0.10) | Complior |
| Litvin et al. [53] | 44 pt with severe OSA and grade II-III HT | Randomized, double-blind, crossover | 6 weeks | −0.7 (−0.95, −0.45) | Sphygmocor |
| Jones et al. [54] | 53 pt with moderate OSA and no CVD, HT or DM | Randomized, double-blind, crossover | 24 weeks | −0.1 (−0.24, 0.04) | Micromanometer (Millar Instruments) and Sphygmocor |
| Paz y Mar et al. [55] | 153 pt with moderate OSA, two arms balanced for cardiopathy, medications and HT | Randomized, double-blind, parallel-group | 2 months | −0.18 (−1.18, 0.82) | Sphygmocor |
| Cardoso et al. [56] | 125 patients with resistant HT | Randomized, not-blinded, parallel-group | 6 months | −0.40 (−0.82, 0.02) | Complior |
Summary of RCTs exploring the effect of continuous positive air pressure (c-PAP) treatment on arterial stiffness measured as carotid-femoral pulse wave velocity (cfPWV)
In one study [52], 24 patients with recently diagnosed OSA, without other comorbidities or CV factors, were randomly assigned to receive c-PAP or no treatment. After 4 months, the treated group had a significant decrease in cfPWV (from 10.4 ± 1.0 to 9.3 ± 0.9 m/s; p = 0.001) while the control group showed no change. Diastolic BP decreased in parallel with arterial stiffness after c-PAP treatment, although the difference was reported not to be significant between the two groups and the change in BP did not correlate with changes in vascular parameters.
In a crossover study of 44 subjects with severe OSA and grade 2 and 3 HT, c-PAP or sham treatments were performed for 3 weeks [53]. On c-PAP, cfPWV showed an additional reduction of 0.7 m/s (p = 0.03) alongside decrease in all BP components including aortic systolic and diastolic BP (approximately 7 and 5 mmHg respectively). Whereas Jones et al. [54] failed to show a significant improvement in vascular stiffness with c-PAP. 53 subjects with moderate-severe OSA (AHI ≥ 15) were randomly assigned to 12-weeks treatment of c-PAP (n = 25) or sham c-PAP (n = 27) before crossing into the other arm of the study for a further 12 weeks. While BP was lowered by c-PAP treatment, there was no difference in cfPWV [54].
In 153 patients with recently diagnosed moderate OSA (AHI ≥ 15) was designed to randomly assign participants to c-PAP treatment (n = 76) or placebo c-PAP (n = 77) for 2 months [55]. In the first group, only 29 subjects were compliant with therapy, while 38 withdrew during the intervention. At the end of the trial, there was no significant difference between cfPWV in the c-PAP group compared to the control group, and neither arm showed BP changes during the trial [55].
Finally, Cardoso et al. [56] conducted a RCT in subjects with resistant hypertension and moderate/severe OSA in which 62 were assigned to cPAP treatment and 63 were control for 6 months. The study did not find any significant reduction in cf-PWV in the interventional group compared to the control one but a differences in the progression of aortic stiffness between the two (−0.40 m/s, 95% CI −0.82 to +0.02 m/s; p = 0.059 mostly driven by a significant increase in arterial stiffness in the control group).
Clearly, the larger trial [55] influenced the most on overall results but had major dropouts and the trial in resistant HT [56] was underpower to show intergroup differences. Interestingly, RCTs which showed a significant decrease in cfPWV (or its progression) included patients with a more severe form of OSA (AHI ≥ 30) and a significantly higher percentage of hypoxia (calculated as sleep time <90% oxygen saturation in total sleep time). This observation is in line with evidence from observational studies and RCTs which studied the effect of c-PAP on BP [57–59]. Another aspect which should be considered is the baseline value of cfPWV which differed in the four trials, with higher values in positive trials (10.4, 13.9 and 9.4 m/s) [52,53] compared to the negative ones (7.6 and 9.05 m/s) [54,55] suggesting a potential limitation related with the effect size. A relatively small sample size, presence of comorbidities and compliance to treatment may also be additional confounders in the analysis.
Of note, none of the RCTs included any measurement of autonomic function which could represent one of the pathophysiological mechanisms linking OSA and arterial stiffness.
To summarize, a large body of evidence supports the role of OSA as a risk factor for increased arterial stiffness. Several pathophysiological mechanisms, including the activation of the autonomic nervous system, may help explain the link between the breathing disorder and the vascular alterations (here mainly examined as functional properties rather than remodelling). Whether a causal relationship between OSA and vascular damage exists or is mostly explained by confounders and whether OSA treatment can improve vascular stiffening is still debated. More RCTs are warranted and their design should consider changes in BP as one of the main confounders in the analysis.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
All authors developed the review concept and designed the layout. DM, LF and PJC wrote the majority of the manuscript. DM, BF and RM conducted the electronic searches and extraction. All authors read, critically evaluated and approved the final version of the manuscript.
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - D. Marcon AU - L. Faconti AU - B. Farukh AU - R. McNally AU - C. Fava AU - M. Pengo AU - P. Chowienczyk AU - J.K. Cruickshank PY - 2020 DA - 2020/11/13 TI - Does Treatment for Obstructive Sleep Apnoea Improve Arterial Stiffness? Evidence from Randomized Clinical Trials on Carotid-femoral Pulse Wave Velocity JO - Artery Research SP - 1 EP - 6 VL - 27 IS - 1 SN - 1876-4401 UR - https://doi.org/10.2991/artres.k.201102.004 DO - 10.2991/artres.k.201102.004 ID - Marcon2020 ER -