
Complex vasoactivity of liraglutide. Contribution of three gasotransmitters
- DOI
- 10.1016/j.artres.2015.04.001How to use a DOI?
- Keywords
- Aortic rings; Central (aortic) vasodilatation; GLP-1; Liraglutide
- Abstract
Background: Incretine hormone glucagon-like peptide-1 (GLP-1) causes dose-dependent relaxation of the thoracic aorta of rats and other arteries via nitric oxide (NO), cAMP and ATP-sensitive potassium channels, however, through a mechanism not thoroughly described. Hereby we aimed to determine the mediators involved in the vasoactive effect of liraglutide.
Methods: Isolated rat aortic rings and segments of the femoral artery were mounted in a wire myograph to study the vasoactive effect of liraglutide. Vessels were preincubated either with inhibitors of gasotransmitter-, prostaglandin- or reactive oxygen species-formation, or with inhibitors of protein kinases, potassium channels or the Na+/Ca2+-exchanger.
Results: According to our findings, liraglutide activates endothelial cells and vascular smooth muscle cells leading to the production of NO, carbon monoxide, hydrogen sulphide, superoxide anion, and hydrogen peroxide. Increased production of such relaxing factors promotes the activation of protein kinase– A and –G, resulting in the activation of potassium channels (ATP-sensitive-, voltage-gated-, large-conductance-calcium activated), which profoundly contributes to the activation of the Na+/Ca2+-exchanger, thereby leading to calcium efflux and smooth muscle relaxation and vasorelaxation.
Conclusions: We reveal the contribution of all gasotransmitters in the vasorelaxation induced by liraglutide. We provide ex vivo evidence that liraglutide is capable of causing vasodilatation in the central and peripherial vessels, thereby supporting the clinical observation that it lowers blood pressure.
- Copyright
- © 2015 Association for Research into Arterial Structure and Physiology. Published by Elsevier B.V. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Introduction
Liraglutide, a glucagon-like peptide-1 (GLP-1) analogue is a drug used in the therapy of type 2 diabetes.1 It exerts its effects mainly via the GLP-1 receptor (GLP-1R), although GLP-1R-independent effects have also been described.1 GLP-1R is expressed, among others, on endothelial cells and vascular smooth muscle cells.2 A broad expression of GLP-1R mRNA in the thoracic aorta of rats has been identified.3
Numerous publications pointed out that native GLP-1 causes concentration-dependent relaxation of different arteries; however, controversial data have been published about the mechanism of the vasodilator effect of GLP-1 and its analogues.3–7 GLP-1 exerted dose-dependent vasorelaxation in the pulmonary arteries of rats in an endothelium-dependent manner,4,5 while GLP-1 causing vasodilatation of the rat femoral artery was found to be endothelium-independent.6 These findings may indicate that GLP-1 evokes vasodilatation via different pathways in the different parts of the arterial system. GLP-1 is known to increase the plasma level of the potent vasodilator nitric oxide (NO).7 Liraglutide induces NO-production in vascular endothelial cells,8 however so far there has been no evidence, that any of the GLP-1 analogues would be able to induce the synthesis of other gasotransmitters.
Potassium channels and protein kinases might be targets of gasotransmitters and other mediators of vasorelaxation.9 GLP-1 and other related peptides were found to cause vasodilatation of the rat thoracic aorta with the contribution of ATP-sensitive potassium channels (KATP) and they are also known to influence the activity of the voltage-dependent potassium channels (Kv).3,10,11
In an ApoE−/− deficient mouse model, liraglutide was shown to have non-PKA, GLP-1R dependent effects in the regulation of eNOS (endothelial nitric oxide synthase) enzyme expression and attenuated intracellular adhesion molecule-I (ICAM-1) expression in aortic endothelial cells, referring to the role of liraglutide in the inhibition of endothelial cell dysfunction.13
Moreover, an increased cAMP production was found in aortic tissue incubated with GLP-1.3
A number of studies demonstrated that both native GLP-1 and its mimetics induce vasodilation, furthermore, some studies focused on their effect in the thoracic aorta, however, the precise mechanism of vasodilatation still remains unclear.3,18
A previous study, which demonstrated the GLP-1R dependent, NO-independent systolic blood pressure lowering effect of liraglutide, also reported that the antihypertensive effect of liraglutide evoked due to the increased secretion of the atrial natriuretic peptide (ANP).12
Central (aortic) blood pressure, the pressure measured in the aorta, is a major determinant of cardiovascular outcomes,14 therefore, the possible effects of liraglutide to lower the pressure in the aorta as well as in other arteries could be beneficial in clinical practice.
Considering the wide diversity of the data mentioned above, we concluded that the precise mechanism of vasodilatation caused by GLP-1 analogue liraglutide is not thoroughly described, thereby in our study we aimed to determine whether liraglutide relaxes the rat thoracic aorta and identify mediators and second messengers involved in the vasodilator effect of liraglutide. Therefore, we studied the effect of liraglutide on gasotransmitters and ion channels.
Materials and methods
Chemicals
Liraglutide (Victoza® injection) was purchased from Novo Nordisk Hungary (Budapest, Hungary). Exenatide (Byetta® injection) was purchased from Bristol-Myers Squibb–AstraZeneca (Budapest, Hungary). Acetylcholine, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), Nω-Nitro-
Animals
All experiments were approved by the Hungarian Local Animal Experiment Committee, in accordance with the ‘Principles of laboratory animal care’ (NIH publication no. 85–23, revised 1985). Animals were originally purchased from Charles River Laboratories GmbH (Sulzfeld, Germany). Adult, 10–12 week old male Sprague-Dawly rats weighing between 280 and 340 g were kept on a standard diet. On the day of the experiment they were anaesthetized with ether and decapitated by a guillotine.
Vasoreactivity experiments
The thoracic aorta and femoral artery of each rat were gently removed and placed in oxygenated (95% O2/5% CO2), ice cold Krebs solution (119 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 25 mM NaHCO3, 1.2 mM Mg2SO4, 11.1 mM glucose, 1.6 mM CaCl2*2H2O, pH 7.4). After removing the perivascular fat and connective tissue as described earlier by Köhn et al.,15 vessels were dissected into 2 mm long segments and mounted on two stainless steel wires (40 μm in diameter) and placed in 5 ml organ baths of a wire myograph (Danish Multimyograph Model 610M, DMT– USA In., Atlanta, GA, USA). Vessels were bathed in Krebs solution at 37 °C, pH 7.4 and were continuously oxygenated with 95% O2, 5% CO2. After 30 min of equilibration, the aortic rings were placed under tension of 1 g, while in the case of femoral artery segments the modified method of Fésüs et al. was used.3,16 Rings were allowed to rest for 30 min, isometric tension was continuously recorded. The rings were preconstricted with 100 nM epinephrine as described earlier,17,18 and this concentration of epinephrine caused 60% contraction force of the 60 mM KCl contraction.17 After reaching a stable contraction plateau, relaxant response to increasing doses of liraglutide was assessed. To compare the vasoactivity of the two GLP-1 analogues, in one set of experiments we applied increasing doses of exenatide to the preconstricted aortic rings. The rate of relaxation caused by liraglutide and exenatide is expressed as the percentage of the contraction evoked by epinephrine. The dosage of liraglutide that we used to relax the aorta correlates well with the dosage of epinephrine we used to preconstrict the vessel. The level of plasma epinephrine is 30 pM at rest,19 while in our experiments we used 100 nM, which is a 3000 times higher concentration.18 Accordingly, the plasma level of liraglutide is 10 nM and in our experiments we used a 3000 times higher concentration.20 In rat femoral artery we found that the same dosage of liraglutide causes greater vasodilatation (see supplemental material), and according to our experiments the same applies to the renal artery (data not shown). The plasma exenatide level was found to be 70 pM,21 while we used a 4500 times higher concentration.18
To identify the extracellular and intracellular mediators of the vasodilator effect of liraglutide, we performed a series of experiments (n = 5, each experiment). Before getting the epinephrine-induced contraction plateau we preincubated the aortic-segments and segments of the femoral artery with different materials. In one set of experiments we mechanically removed the endothelium of the vessels by gently rubbing a piece of hair through it. The effect of denudation was verified by the loss of response to 3 μM acetylcholine. We incubated one group of vessels with the eNOS inhibitor L-NAME (300 μM, 30 min). Other vessels were incubated with the potent heme oxygenase inhibitor Tin-protoporphyrin IX dichloride (10 μM, 30min) to block carbon monoxide (CO) formation, others with
Untreated time-control experiments were carried out to exclude the effect of the spontaneous relaxation of the vessel. In order to determine the effect of the above mentioned specific inhibitors on the permanence of the epinephrine-induced plateau, time-control experiments of the inhibitor studies were performed. Since most of the chemicals had only a slight vasodilator effect, this could not significantly influence the results.
The software Myodaq 2.01 M610+ was used for data acquisition and display.
Statistics
Statistical analyses were performed by using SPSS Version 19.0 (SPSS Inc., Chicago, IL, USA) and GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, CA, USA). Statistical significance was calculated using Student’s t-test or ANOVA with Bonferroni post hoc test as appropriate. Values are given as a mean ± SE. A value of P less than 0.05 was considered to be significant.
Results
Liraglutide causes dose-dependent vasorelaxation
To start with, we evaluated the spontaneous relaxation of the vessel precontracted with epinephrine, however untreated vessels showed no significant spontaneous relaxation. (Fig. 1A) When assessing the vasoactive effect of this GLP-1 analogue, we administered increasing doses of liraglutide to the vessel chamber after its precontraction with epinephrine. In our experiment we showed that liraglutide dose-dependently relaxed the rat thoracic aorta and the femoral artery, however, respective concentrations of liraglutide elicited greater vasodilation in the femoral artery than in the thoracic aorta (Fig. 1B, Table 1 and supplemental material).
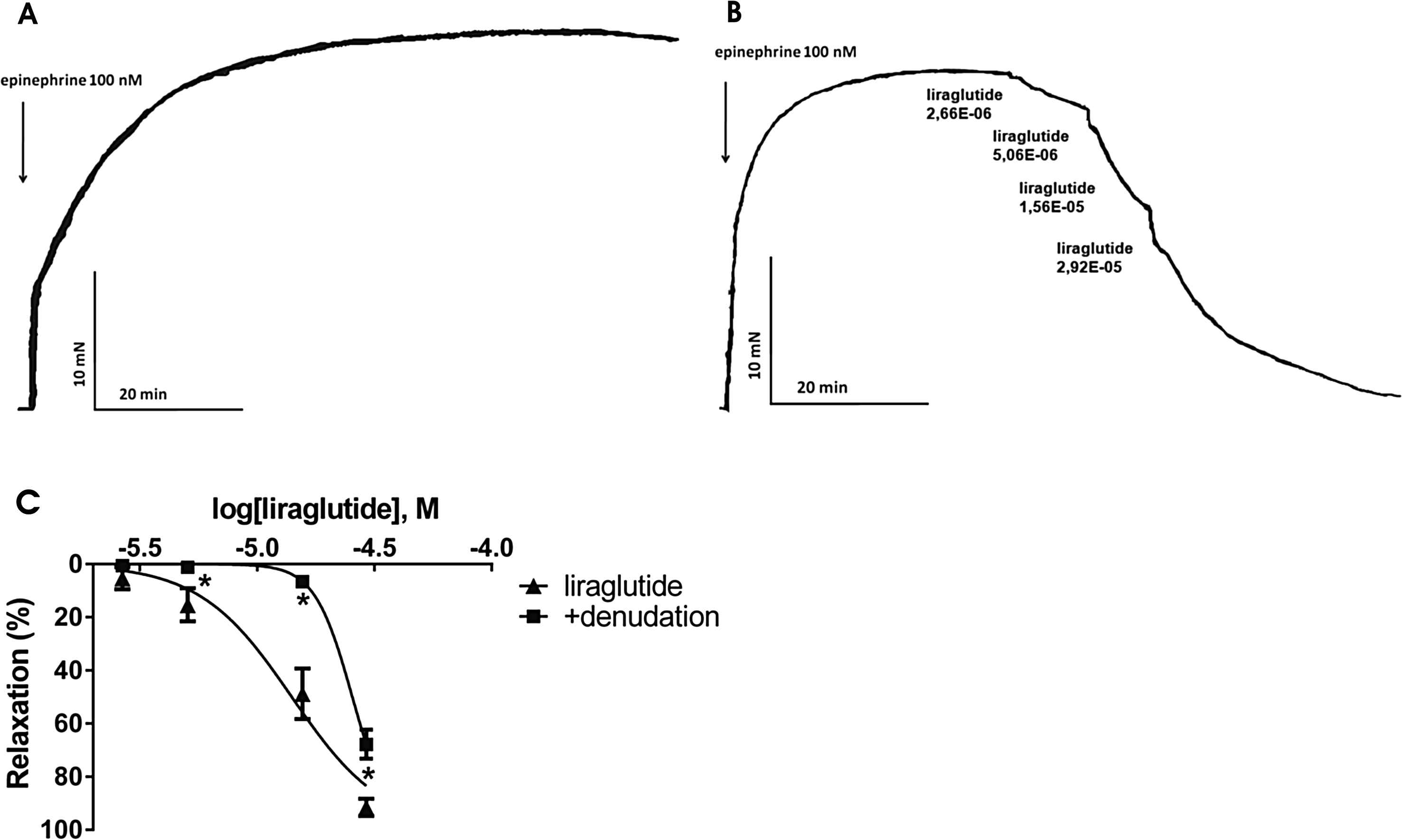
A: Original record, a time-control of an epinephrine precontracted aortic ring. B: Original record of the response of an isolated thoracic aorta segment to 100 nM epinephrine followed by increasing doses of liraglutide (n = 5). C: Liraglutide concentration–relaxation curves in endothelium-intact (▴) and endothelium-denuded (■) vessels (n = 5). *P < 0.001 compared to the relaxation caused by liraglutide only (at respective concentration of liraglutide).
| log EC50 of liraglutide | p | ||
|---|---|---|---|
| Thoracic aorta | Femoral artery | ||
| liraglutide alone | −4.89(0.04) | −5.11(0.10) | 0.004 |
| liraglutide + | |||
| denudation | −4.58(0.02)* | −4.90(0.17) | 0.004 |
| L-NAME | −4.40(0.05)* | −5.36(0.18)* | 0.001 |
| Tin-PP | −4.59(0.01)* | −5.05(0.15) | 0.016 |
| PPG | −4.48(0.16)* | −4.71(0.21)** | 0.200 |
| indomethacin | −4.49(0.09)* | −5.10(0.21) | 0.029 |
| SOD | −4.30(0.01)* | −4.99(0.16) | 0.029 |
| catalase | −3.67(0.96)* | −5.21(0.15)* | 0.010 |
| H89 | −4.18(0.19)* | −4.65(0.08)** | 0.029 |
| ODQ | −4.27(0.29)* | −4.80(0.13)** | 0.029 |
| TEA | −4.27(0.08)* | −4.48(0.20)** | 0.343 |
| glibenclamide | −4.37(0.08)* | −5.08(0.34) | 0.016 |
| XE991 | −3.88(0.15)* | −4.72(0.18)* | 0.100 |
| SEA0400 | −3.52(0.75)* | −4.63(0.02)** | 0.029 |
Abbreviations: L-NAME, Nω-Nitro-
p < 0.05 vs. log EC50 of liraglutide alone in the same vessel segment
p < 0.01 vs. log EC50 of liraglutide alone in the same vessel segment.
Log EC50 values of liraglutide induced vasodilation in the persence of different inhibitors in rat thoracic aorta and femoral artery.
Vasoactive effect of exenatide on the rat thoracic aorta
Exenatide also caused dose-dependent relaxation of the thoracic aorta, although lower concentrations of exenatide were needed to induce vasodilatation (see supplemental material).
Effect of endothelial denudation
Mechanical removal of the endothelium of the thoracic aorta caused a mild inhibition of the relaxation caused by liraglutide. However, this difference in the relaxation between endothelium-intact and endothelium-denuded vessels proved to be significant except for the lowest dosage of liraglutide (Fig. 1C). In the femoral artery, endothelial denudation caused no change in the vasodilator response to liraglutide (Table 1).
Role of gasotransmitters in vasodilatation caused by liraglutide
We incubated a group of vessels with L-NAME, and found that inhibition of eNOS partially avoids vasodilatation caused by liraglutide in both arteries. Decrease in the magnitude of relaxation proved to be significant (Fig. 2A, Table 1).
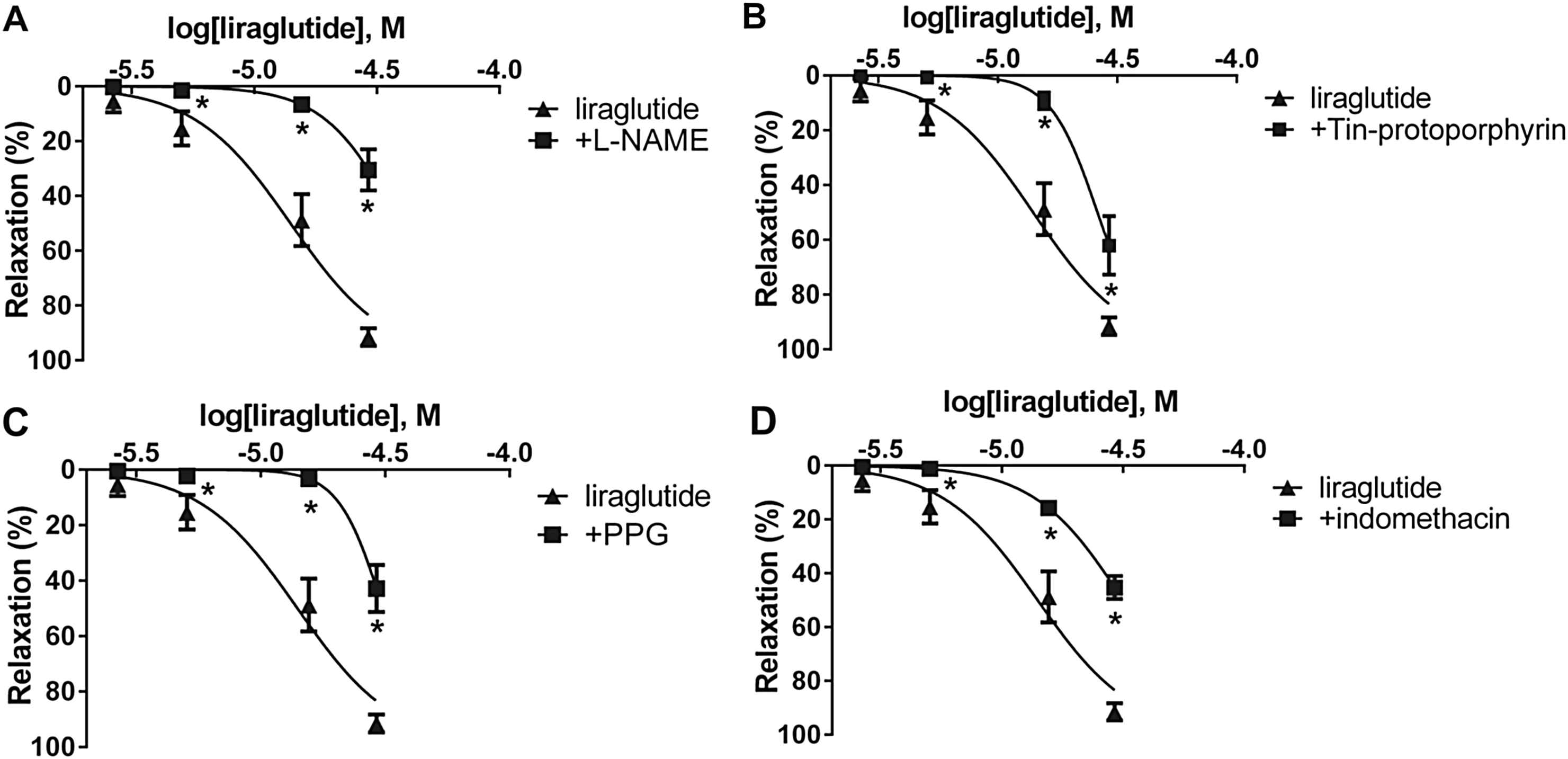
Concentration-relaxation curves of liraglutide (▴) and liraglutide + inhibitors of the potential mediators of the vasodilatation caused by liraglutide (■). A: Inhibition of NOS (Nitric oxide synthase) with 300 μM Nω-Nitro-
To determine further mediators of the vasodilator effect of liraglutide, we tested the role of CO. We preincubated vessels with Tin-protoporphyrin, which is a potent inhibitor of heme oxygenase. Inhibition of CO-production resulted in a significantly milder relaxation in the thoracic aorta, but it had no significant effect in the femoral artery (Fig. 2B, Table 1).
Because the inhibition of NO-synthesis and the inhibition of CO-production only partially decreased the vasodilatation, we wanted to determine whether H2S also plays a part in the vasoactive effect of liraglutide. We inhibited cystathionine-γ-lyase by preincubating vessels with PPG, which significantly decreased the relaxation in both vessels (Fig. 2C, Table 1).
Effect of indomethacin, a prostaglandin synthesis inhibitor
Incubation of vessels with the COX inhibitor indomethacin for 30 min caused significantly smaller vasodilatation in response to liraglutide in the thoracic aorta, but it had no effect in the femoral artery (Fig. 2D, Table 1).
Free radicals contribute to the effect of liraglutide
In order to evaluate the role of ROS in the vasorelaxation caused by liraglutide, we preincubated vessels with superoxide dismutase or with catalase. We found significant inhibition of relaxation in both experiments in case of the aorta, however, we proved that in the femoral artery only H2O2 is involved in the liraglutide induced vasorelaxation (Fig. 3, Table 1).
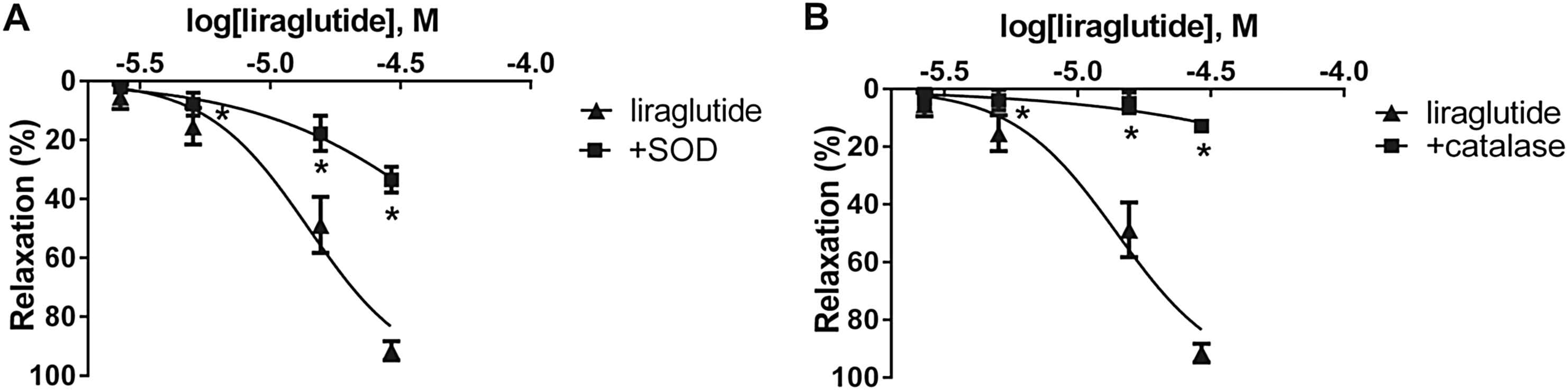
Role of free radicals in the effect of liraglutide. A: Concentration-relaxation curve of liraglutide +200 U/ml of the free radical scavenger superoxid dismutase (SOD) (n = 5). B: Concentration-relaxation curve showing the possible role of hydrogen peroxide by blocking its formation with 1000 U/ml catalase (n = 5). *P < 0.001 compared to the relaxation caused by liraglutide only (at respective concentration of liraglutide).
The role of cAMP-dependent protein kinase A and cGMP-dependent protein kinase G
To determine the second mediator of the dilation caused by liraglutide, we incubated vessels with H89, the inhibitor of PKA. This almost completely abolished the relaxation induced by liraglutide in both arteries (Fig. 4A, Table 1). We inhibited soluble guanylyl cyclase by incubating vessels with ODQ. This led to a significant inhibition of vasorelaxation in both cases (Fig. 4B, Table 1).
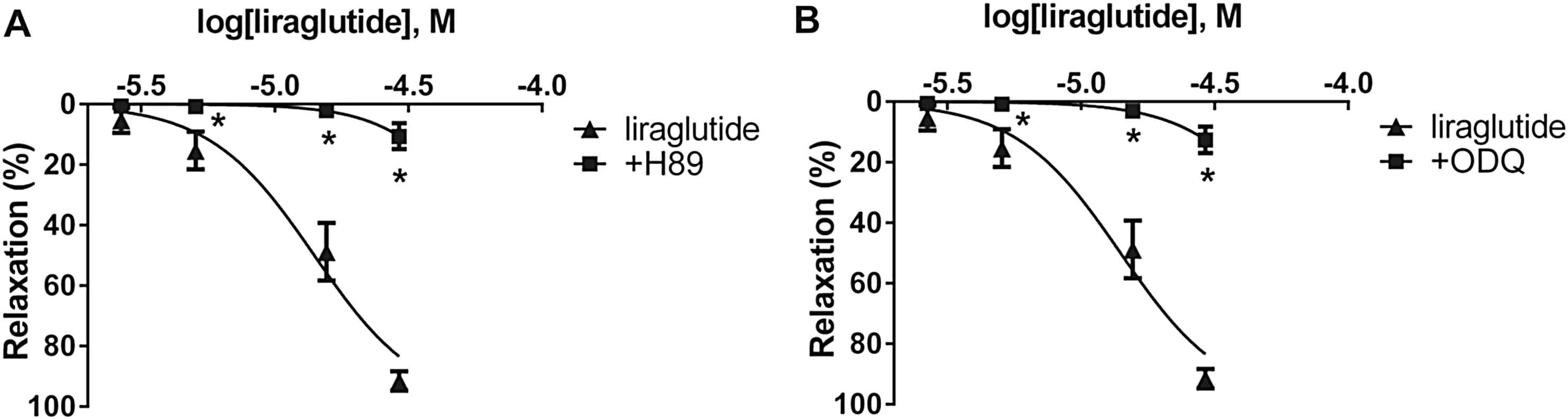
Concentration-relaxation curves showing the possible effector molecules of the liraglutide induced vasodilatation. A: Blocking cAMP-dependent protein kinase A (PKA) with 5 μM H89 hydrochloride (n = 5). B: Inhibition of soluble guanylyl cyclase with 3 μM 1H-[1, 2, 4] oxadiazolo[4,3-a] quinoxalin-1-one (ODQ) (n = 5). *P < 0.001 compared to the relaxation caused by liraglutide only (at respective concentration of liraglutide).
Inhibition of potassium channels with TEA, glibenclamide and XE991
Incubating one group of vessels with TEA, inhibitor of the BKCa channels proved to be a very potent inhibitor of vasodilatation (Fig. 5A, Table 1). Blockade of the KATP channels by preincubation with glibenclamide also inhibited most of the aortic vasodilatation (Fig. 5B). We preincubated one group of vessels with XE991, a KCNQ channel (a type of Kv channels) inhibitor, which also inhibited the vasodilatation in both arteries (Fig. 5C, Table 1).
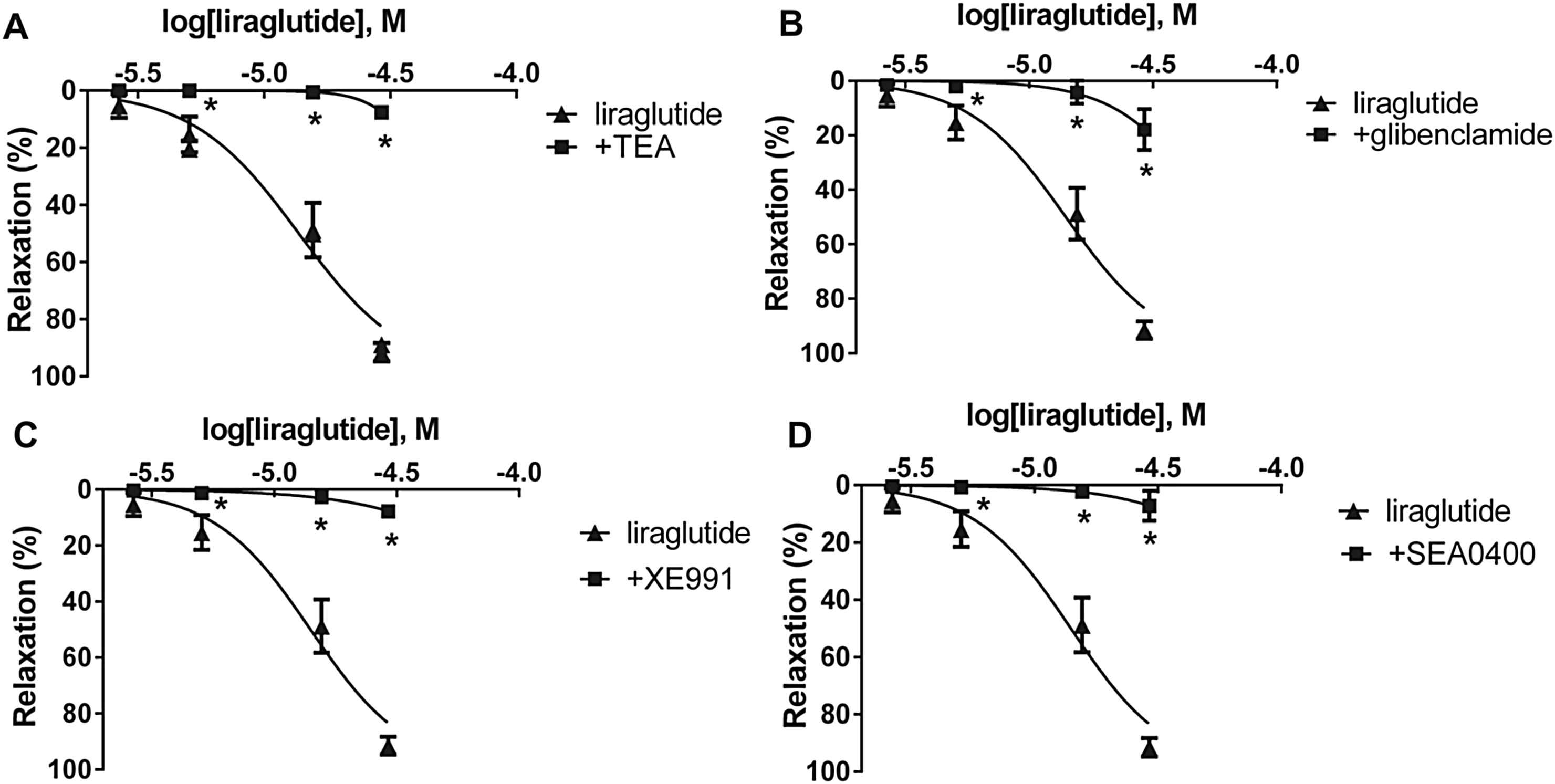
Liraglutide concentration–relaxation curves with (■) and without (▴) an inhibitor of a possible way of the vasodilator effect. A: blockade of large-conductance calcium-activated potassium channels with 2 mM tetraethylammonium (TEA) (n = 5). B: inhibition of ATP-sensitive potassium channels with 10 μM glibenclamide (n = 5). C: KCNQ-type Kv channels blocked by 30 μM XE991 (n = 5). D: Selective inhibition of the Na+/Ca2+-exchanger with 4 μM SEA0400 (n = 5). *P < 0.001 compared to the relaxation caused by liraglutide only (at respective concentration of liraglutide).
Discussion
In our study liraglutide caused dose-dependent relaxation of the rat thoracic aorta and the femoral artery, and it proved to be a more potent vasodilator in the femoral artery.
Our hypothesis for the mechanism of vasorelaxation caused by liraglutide in the thoracic aorta according to our novel findings is as follows: liraglutide activates endothelial cells and vascular smooth muscle cells resulting in an increased production of NO, CO, H2S,
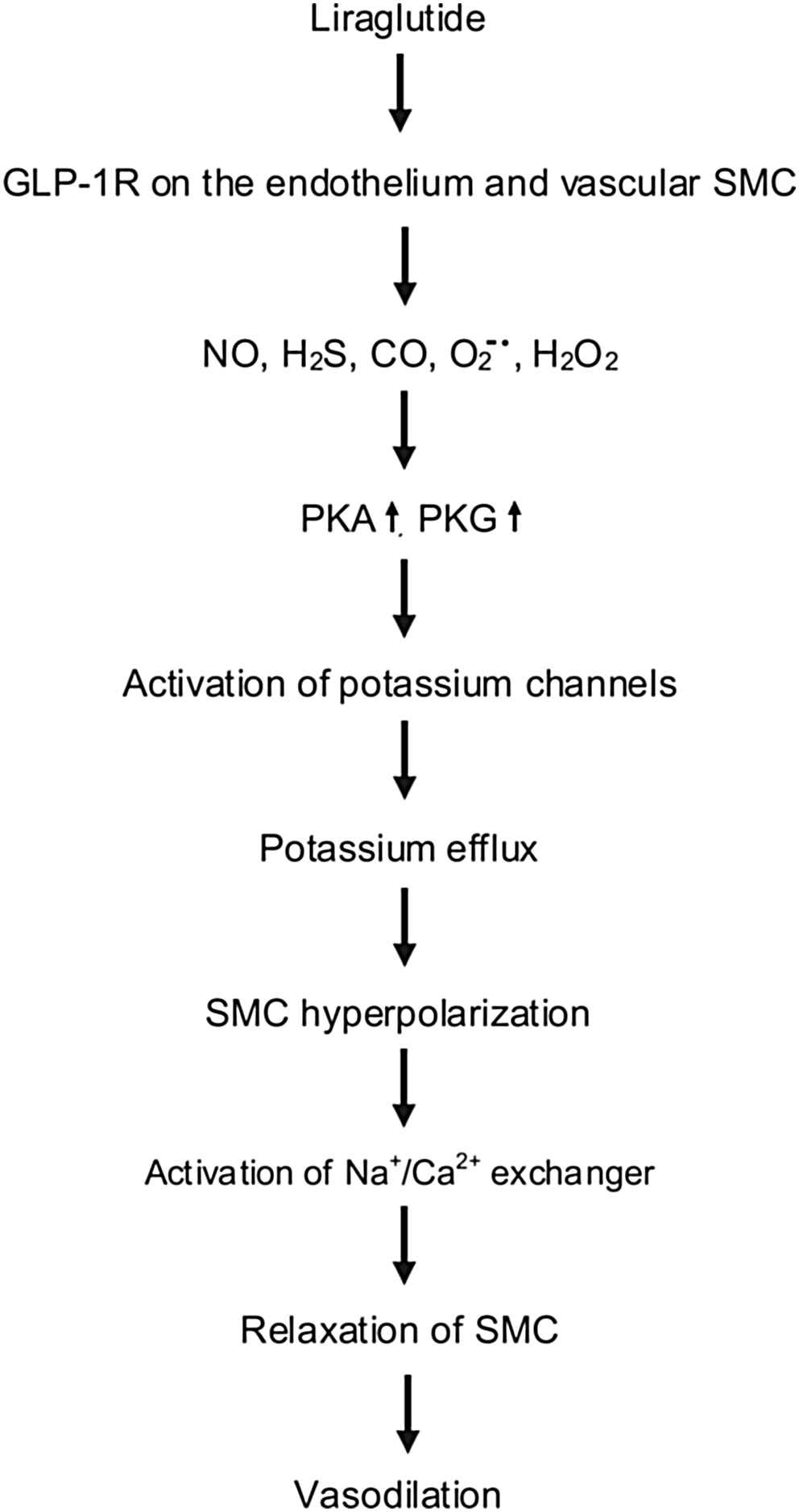
Hypothetical mechanism of liraglutide induced vasodilatation. NO: nitric oxide, H2S: hydrogen sulfide, CO: carbon monoxide,
In contrast to the aorta, in the femoral artery the vasodilation was found to be independent of the endothelium, however partially mediated by NO, H2S and H2O2, while PKA and sGC were both involved, activating the calcium-activated and the KCNQ-type voltage-gated potassium channels, resulting in vasodilation via the activation of the Na+/Ca2+-exchanger.
Despite the number of studies supporting the fact that the vasodilator effect of GLP-1 and its mimetics is mediated via NO,4,5,7 another workgroup found that the vasodilatation evoked by native GLP-1 was independent of NO, since endothelial denudation and incubation with L-NAME had no effect on the relaxation raised by GLP-1 in rat thoracic aorta.3 In concordance with the latter, in our experiments in the isolated rat thoracic aorta, inhibition of eNOS and endothelial denudation only partially inhibited relaxation. This suggests that other mediators (CO and H2S) also take part in the vasodilatation induced by liraglutide.
CO is another significant regulator of vascular tone.24 Like NO, CO is a vasodilator gaseous molecule produced either in the endothelium or in vascular smooth muscle cells from heme by heme oxygenase (HO).9 One vasodilator pathway of CO is the activation of the voltage-dependent potassium channels (Kv).25 A relationship between GLP-1-effects and CO has not been described earlier, however, in our study we demonstrated that CO significantly contributes to the vasorelaxation caused by liraglutide in the thoracic aorta.
The third influential gasotransmitter, H2S is also known to relax blood vessels.9 It is produced in endothelial cells and vascular smooth muscle cells from homocystein, cysthationine or
A previous study found that eNOS inhibitor L-NAME, COX-inhibitor indomethacin, and H2O2 scavenger catalase had no significant effect on the relaxation evoked by native GLP-1 in rat thoracic aorta.3 On the contrary, we found that prostaglandins, NO, H2O2 and
In some arteries, reactive oxygen species (ROS) cause vasodilatation.32 ROS were also found to relax both pulmonary and mesenteric arteries.33,34 Hydrogen peroxide (H2O2) activates Kv channels, thereby causing dilation of the arteries.33,34 H2O2 has previously been found to induce both endothelium-dependent and endothelium-independent relaxation in the rabbit’s aorta.32 Our results suggest, that H2O2 contributes to the liraglutide induced vasorelaxation in the femoral artery and thoracic aorta.
The possible explanation for the discrepancies between the above mentioned studies and ours could be, that while in former studies the endogenous, physiological isoform GLP-1(7–36) amide was used, we studied the effects of the GLP-1 analogue liraglutide.
Our findings suggest the involvement of all three gasotransmitters in the vasoactive effect of liraglutide, which has not been described earlier. A possible explanation of this might be that former studies focused mainly on the role of NO, and showed less interest in a detailed description of the mechanism of vasorelaxation caused by liraglutide.
Activation of PKA by liraglutide has been described many times, but these studies did not mention the involvement of PKG.10,11 However, we found that both PKA and PKG are involved in the vasoactive effect of liraglutide.
The role of potassium channels in the effects of GLP-1 on β-cells has thoroughly been described,10,11 however, in the vasodilator effect of GLP-1 only KATP channels have been shown to express a proven role.3 KATP channels are expressed on vascular smooth muscle cells, and they are responsible for vasodilatation in many cases.22 A former study demonstrated that GLP-1 causes vasodilatation of the rat thoracic aorta via the activation of KATP channels.3 Unlike GLP-1, sulphonylurea drugs are known inhibitors of vasodilatation via KATP channels, thereby when liraglutide and a sulphonylurea are applied simultaneously for the treatment of type 2 diabetes, the sulphonylurea component might inhibit the vasodilator effect of liraglutide.35
The vasorelaxation caused by H2S, which, as we have demonstrated, is involved in the vasorelaxation evoked by liraglutide often arises via the activation of KATP and KCNQ-type Kv channels.
We have found that both KATP and KCNQ-type Kv channels are activated by liraglutide in the vasculature, which leads to vasodilatation. Both carbon monoxide and superoxide are known activators of these channels.22
According to our findings, another type of potassium channels, the large conductance calcium activated potassium channels are also involved in vasodilatation due to liraglutide. There has been no evidence for the role of BKCa channels in GLP-1 activity so far, although they are constantly expressed in vascular smooth muscle cells.22 The vasodilator effect of CO partially depends on the activation of these channels, moreover, the activation of PKG by NO or the activation of PKA by its mediators result in the activation of these channels.22,26
The sodium–calcium exchanger (Na+/Ca2+-exchanger or NCX) is a transmembrane protein found almost in all cell types.36,37 It is responsible for calcium efflux and sodium influx in hyperpolarization, whereas when the cell membrane is depolarized, its activity is reversed and it induces calcium influx.36,37 According to our results, the NCX might be the terminal effector in vasodilatation caused by liraglutide in the rat thoracic aorta and the femoral artery.
We provide ex vivo evidence for the mechanism of the liraglutide induced vasorelaxation in the thoracic aorta and in the femoral artery. We have also proved that liraglutide is a more potent vasodilator in the femoral artery, speculating that liraglutide may reduce the arterial stiffness gradient, which could be highly beneficial in clinical practice.
However, the specificity of the inhibitors and the usage of a single methodology (myography) undoubtedly limit the relevance of our findings.
Conclusion
To summarize our findings, liraglutide induces vasodilation in both central and peripherial vessels, thus according to our findings the vasodilator effect of liraglutide was induced by three gasotransmitters (NO, CO, H2S) and also by reactive oxygen species and prostaglandins via activation of PKA and PKG, which involves the activation of several potassium channels and the Na+/Ca2+-exchanger. This mechanism is far more diverse than it was assumed earlier.
Disclosure
The authors declare that they have no conflict of interest.
Acknowledgements
The present scientific contribution is dedicated to the 650th anniversary of the foundation of the University of Pécs, Hungary. This research was supported by the European Union and the State of Hungary, co-financed by the European Social Fund in the framework of TÁMOP 4.2.4. A/2-11-1-2012-0001 ‘National Excellence Program’. The authors express their sincere thanks to Ildikó Fábián (University of Pécs, Department of Surgical Reserch and Techniques) and Krisztina Szalma (University of Pécs, 2nd Department of Internal Medicine and Nephrological Center) for their outstanding technical assistance.
Appendix A.
Supplementary data
Supplementary data related to this article can be found at
References
Cite this article
TY - JOUR AU - Eszter Sélley AU - Gergő A. Molnár AU - Szilárd Kun AU - István András Szijártó AU - Boglárka Laczy AU - Tibor Kovács AU - Ferenc Fülöp AU - István Wittmann PY - 2015 DA - 2015/05/09 TI - Complex vasoactivity of liraglutide. Contribution of three gasotransmitters JO - Artery Research SP - 1 EP - 9 VL - 11 IS - C SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2015.04.001 DO - 10.1016/j.artres.2015.04.001 ID - Sélley2015 ER -