
Prostaglandin E1 for preventing the progress of pulmonary arterial hypertension in rat model
- DOI
- 10.1016/j.artres.2018.05.001How to use a DOI?
- Keywords
- Heart; Lung; Prostaglandin E1; Pulmonary artery hypertension; Right ventricular
- Abstract
Pulmonary artery hypertension (PAH) is a progressive chronic disease with a high mortality rate. Increased pulmonary vascular resistance and over-proliferation of pulmonary artery endothelial cells lead to remodeling of pulmonary vasculature. Several anti-PAH therapies targeting various pathways involved in PAH progression have been approved by the FDA. However, many of the currently available anti-PAH drugs suffer from a number of limitations, including short biological half-life, and poor pulmonary selectivity. Prostaglandin E1 (PGE1) is a compound with vasodilatory, anti-inflammatory, anti-aggregatory, and anti-proliferative properties. Recently, PGE1 is known to accumulate in sites of inflammation or vascular lesions and thus enhance the effects of the drugs and alleviate the side effects. Therefore, we hypothesized that long-term effect of PGE1 could reduce ma adaptive structural remodeling of the lung and heart and prevent ventricular arrhythmias in monocrotaline (MCT)-induced rat model of PAH. Our results revealed that PGE1 reduced ventricular hypertrophy, protein expressions of endothelin-1 (ET-1) and endothelin receptor A (ERA), and the expression of fibrosis. These results support the notion that PGE1 can improve the functional properties of RV, highlighting its potential benefits for heart and lung impairment.
- Copyright
- © 2018 Association for Research into Arterial Structure and Physiology. Published by Elsevier B.V. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Introduction
Pulmonary artery hypertension (PAH) is a progressive chronic disease with a high mortality rate.1 Its cardinal signs include an elevation of pulmonary artery pressure, right ventricular (RV) hypertrophy, and arteriolar wall remodeling.2 Increased pulmonary vascular resistance and over-proliferation of pulmonary artery endothelial cells lead to remodeling of pulmonary vasculature.3–5 Prostacyclin analogues targeting the abnormal contraction of the pulmonary artery have emerged as one of the promising therapies.6–9 Continuous infusion of epoprostenol, a prostacyclin analogue, was reported to improve cardiac function, oxygen saturation, and exercise capacity in PAH.10
Prostaglandin E1 (PGE1; Alprostadil) is a compound with vasodilatory, anti-inflammatory, anti-aggregatory, and anti-proliferative properties.11–13 It has been reported that PGE1, when administered via the pulmonary route, works as a selective pulmonary vasodilator.14 Recently, PGE1 is known to accumulate in sites of inflammation or vascular lesions and thus enhance the effects of the drugs and alleviate the side effects.15,16 However, the clinical utility of PGE1 has been limited by its chemical instability and extremely hydrophobic nature, which results in low drug absorption and poor bioavailability. And, the main limitation of pulmonary delivery of PGE1 is its very short half-life (3–4 min) as a result of first-pass metabolism in the lungs. It has been demonstrated that PGE1 can improve pulmonary hemodynamics and clinical outcomes of PAH.17 However, the long-term effect of PGE1 on rats with monocrotaline (MCT)-induced PAH has not been investigated. Therefore, the aim of this study was to evaluate the effect of continuous intravenous PGE1 on hemodynamics and long-term outcomes of rats with MCT-induced PAH. To determine changes in pulmonary hemodynamics, right ventricular hypertrophy, pathology, and protein expressions, we used immunohistochemical staining and western blot analysis to understand whether PGE1 treatment could reduce the development of PAH in the model.
Methods
Animals
Six-week-old male Sprague–Dawley rats were used. All rats were housed in climate-controlled conditions with a 12-h light/12-h dark cycle, and had free access to food and water. All animal experiments were approved by the appropriate Institutional Review Boards of the Seoul National University Bundang Hospital (BA1607-205/044-01) and conducted in accordance with National Institutes of Health Guide for the Care Use of Laboratory Animals (NIH publication No. 86-23, revised in 1996).
Pulmonary arterial hypertension (PAH) rat model
PAH was induced by subcutaneous (sc) injection of 60 mg/kg monocrotaline (MCT) (Sigma–Aldrich, St. Louis, MO, USA) dissolved in 0.5 N HCl. Rats were assigned into the following groups: 1) control (C) group (20 rats), vehicle injection with normal diet; 2) monocrotaline group (M) (20 rats), MCT injection with normal diet; and 3) Prostaglandin E1 (PGE1) group (20 rats), MCT injection + PGE1 injection with normal diet. These animals were sacrificed at 7, 14, 21, and 28 days (each group, n = 5) after MCT administration. Tissues were removed and immediately frozen at −70 °C for enzyme analysis.
Drug delivery
Osmotic infusion pump (Alzet Model 2ML-1, ALZA Corp., Palo Alto, CA) was utilized as drug delivery system as described previously.18 Alzet 2ML-1 is a fully implantable and osmotically driven pump. It can deliver 2 ml over a period of 7 days at a constant rate of infusion. Prostaglandin E1 (Lot U-10136, Upjohn, Kalamazoo, MI) was dissolved in absolute ethanol dried over sodium sulfate and added to the pump to yield an infusion rate of 0.1 pg/kg/min. This dose was chosen based on previous experiment showing that animals receiving it could tolerate high doses of PGE1.19 Based on pilot studies, PGE1 was found to be stable in the pump for 7 days at pH 7.4.
Determination of the organ weights and right hypertrophy index
The rats were weighed and observed for general appearance during the study period. The animals were sacrificed at the scheduled time. The wet weights of excised right ventricle (RV), left ventricle (LV) plus interventricular septum (IVS) (LV+IVS) were measured. The RV to LV+IVS ratio [RV/(LV+IVS)] was used to determine the right hypertrophy index (RVI). The standard of right ventricular hypertrophy was defined as an RVI > 0.33.20
Pulmonary hemodynamics
Rats were anaesthetized by intraperitoneal injection of urethane and secured on a surgical stage. An 8-mm-long right internal jugular vein was isolated and ligated at the distal end. The vessel was cut at the proximal end of ligation. A catheter filled with heparin saline was rapidly inserted along the incision and slowly advanced for about 5 cm to enter the pulmonary artery. The standard of pulmonary hypertension was defined as systolic pulmonary artery pressure (SPAP) > 50 mmHg.20 Hemodynamic parameters were recorded at baseline and at 7, 14, 21 and 28 days.
Histologic findings of pulmonary arteries
Heart and Lung tissues were fixed with 10% buffered formalin and then embedded in paraffin. Sections were performed by four micron-thick hematoxylin-eosin stains to evaluate histopathologic changes of pulmonary blood vessels. The small pulmonary artery wall thickness was expressed as follows: % wall thickness.
Masson trichrome staining
Masson trichrome staining was carried out in accordance with well-characterized protocols. Briefly, heart and lung tissue sections were deparaffinized and hydrated in distilled water prior to a 1-hr treatment in Bouin’s fixative (Richard-Allan Scientific; CA, USA; Catalog #NC9674780) at 56 °C. Sections were washed in running distilled water until clear, and then stained in Weigert’s iron hematoxylin (Richard-Allan Scientific; Catalog #NC9231529) for 10 min. Following a 10-min wash in running water, sections were stained in Biebrich scarlet-acid fuchsin (Richard-Allan Scientific; Catalog #NC9424144) for 2 min. Sections were rinsed in distilled water followed by a 10-min differentiation in phosphomolybdic-phosphotungstic acid (Richard-Allan Scientific; Catalog #NC9443038). Aniline blue (Richard-Allan Scientific; Catalog #NC9684104) was used as a counterstain for 10 min, and then sections were differentiated in 1% acetic acid for 3 min. Sections were dehydrated through a series of graded alcohols back to xylene, and then coverslipped and sealed using Cytoseal XYL (Richard-Allan Scientific).
Immunohistochemistry
Excised lung tissues were incubated overnight in 10% buffered formalin. Four-micrometer-thick sections were cut from paraffin embedded tissue blocks, deparaffinized in xylene, and rehydrated in graded alcohol solutions (70%–100%). Heat antigen retrieval was achieved by boiling the tissue sections in antigen retrieval solution in pH 6.0 or pH 9.0 (Dako, Carpinteria, CA, USA) for 10 min in a microwave prior to incubation at 4 °C overnight with primary antibodies against endothelin-1 (ET-1; Abcam, Cambridge, MA, USA). After incubation with the appropriate biotinylated secondary antibodies for 30 min at 4 °C and subsequently with streptavidin (Dako, Kyoto, Japan), color development was done using diaminobenzidine (DAB) as a chromogen and counterstained with hematoxylin.
In situ terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling technique assay for lung cell apoptosis
Apoptotic cells in the lung tissue sections were detected by the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling technique (TUNEL) using a commercial apoptosis kit (TACS TM TdT Kit, R&D Systems), according to the supplier’s instructions. In brief, the lung tissue sections were de-paraffinized with xylene and ethanol and rinsed with PBS. The sections were then treated with proteinase K in PBS followed by quenching of endogenous peroxidase. A biotinylated dNTP mix was added to the 3′OH ends of DNA by terminal deoxynucleotidyl transferase (TdT). After incubating with streptavidin-horseradish peroxidase, the sections were stained with DAB and counterstained with methyl green. Finally, the sections were dehydrated in ethanol, cleared with xylene, and mounted with coverslips in a permanent medium. According to the supplier’s instructions, experimental controls included for the assay were TACS-nuclease–treated thyroid tissue sections as the positive control and the omission of the TdT reaction step as the negative control.
Western blot analysis
The tissue was homogenized in 10 mM Tris HCl buffer, pH 7.4 containing 0.5 mM EDTA, pH 8.0, 0.25 M sucrose, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na4VO3 and a protease inhibitor cocktail (Roche-Boehringer-Mannheim, Mannheim, Germany). After centrifugation, the supernatant was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Samples equivalent to 25 μg of protein content were loaded and size-separated in 8–12% SDS-PAGE. The proteins on the acrylamide gel were transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford MA, USA) at 400 mA in a transfer buffer containing 25 mM Tris and 192 mM glycine, pH 8.4. The nitrocellulose membranes was blocked in tris-buffered saline with 5% non-fat dry milk at room temperature for 1 h in 0.1% Tween 20 and incubated with the appropriated primary antibodies, including anti-ET-1 and anti- ERA (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and anti-actin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), at 4 °C overnight. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology Inc., Danvers, MA, USA) for 1-hr at room temperature. After washing, the membranes were visualized by a chemiluminescent ECL-detection kit from GE-Healthcare (Piscataway NJ, USA).
Statistical analyses
Results were expressed as the mean ± standard deviation. Differences between all other parameters for the three groups were evaluated by ANOVA followed by multiple-group comparisons. An unpaired two-tailed t-test and Mann–Whitney test were used, and a P value < 0.05 was considered statistically significant. SPSS 14.0 for windows (SPSS, Chicago IL, USA) was used for all statistical analyses.
Results
Organ weight
RV weight of the M group was increased at 21 and 28 days after MCT administration. Weights of LV+IVS were not significantly different among C, M, and PGE1 groups. RV/LV+IVS ratios were significantly increased at 14, 21, and 28 days in the M group compared to those in the C group. However, RV/LV+IVS ratios were significantly decreased at 21 and 28 days in the M group compared to those in the PGE1 group. Weights of LV+IVS were significantly lower in the M group and the PGE1 group compared to those in the C group at 21 and 28 days. Heart weights were significantly increased in the M group compared to those in the C group at 21 and 28 days. However, heart weights were significantly decreased in the PGE1 group compared to those in the M group at 21 and 28 days (Table 1).
| Day | Group | SASP (mmHg) | RV (g) | LV + IVS (g) | RV/(LV+IVS) % |
|---|---|---|---|---|---|
| 7 | Control | 21.4 ± 1.3 | 0.142 ± 0.02 | 0.543 ± 0.03 | 0.26 ± 0.15 |
| M | 24.3 ± 2.5 | 0.149 ± 0.03 | 0.552 ± 0.03 | 0.27 ± 0.23 | |
| PGE1 | 23.1 ± 2.3 | 0.148 ± 0.03 | 0.550 ± 0.02 | 0.27 ± 0.18 | |
| 14 | Control | 22.4 ± 1.1 | 0.149 ± 0.03 | 0.658 ± 0.03 | 0.22 ± 0.18 |
| M | 35.2 ± 3.1* | 0.208 ± 0.05 | 0.643 ± 0.04 | 0.32 ± 0.32* | |
| PGE1 | 31.6 ± 2.4 | 0.171 ± 0.04 | 0.664 ± 0.05 | 0.25 ± 0.24 | |
| 21 | Control | 23.7 ± 1.3 | 0.162 ± 0.03 | 0.740 ± 0.04 | 0.21 ± 0.22 |
| M | 48.9 ± 6.1* | 0.306 ± 0.05* | 0.642 ± 0.05 | 0.48 ± 0.53* | |
| PGE1 | 37.7 ± 4.2# | 0.191 ± 0.03 | 0.706 ± 0.05 | 0.27 ± 0.35# | |
| 28 | Control | 24.4 ± 1.2 | 0.171 ± 0.04 | 0.783 ± 0.05 | 0.22 ± 0.24 |
| M | 55.3 ± 9.3* | 0.366 ± 0.10* | 0.682 ± 0.08 | 0.54 ± 0.68* | |
| PGE1 | 39.5 ± 4.3# | 0.212 ± 0.06# | 0.722 ± 0.07 | 0.29 ± 0.35# |
Values are presented as means ± standard deviation. Control, Control group; M, monocrotaline group; PGE1, Prostaglandin E1 group; SPAP, systolic pulmonary artery pressure; RV, right ventricle; LV, left ventricle; IVS, interventricular septum.
P < 0.05 compared with the C group,
P < 0.05 compared with the MCT group.
Changes of SPAP and organ weights after PGE1 treatment in PAH rats.
Changes in systolic pulmonary artery pressure (SPAP) after PGE1 treatment in PAH rats
Mean SPAP values were significantly increased in the M group compared to those in the C group at 14, 21, and 28 days. SPAP values were significantly decreased in the PGE1 group compared to those in the M group at 21 and 28 days (Table 1).
Medial wall thickness of the pulmonary artery
In the M group, the ratio of medial thickening to the external diameter of the pulmonary artery was significantly increased from 28 days compared to that in the control group. In the PGE1 group, the ratio of medial thickening of the pulmonary artery was not significantly decreased compared to that in the M group (Table 2).
| Day | Control | M | PGE1 |
|---|---|---|---|
| 7 | 9.5 ± 0.5 | 8.8 ± 0.3 | 8.9 ± 0.4 |
| 14 | 9.2 ± 1.1 | 12.7 ± 1.2 | 12.1 ± 1.1 |
| 21 | 8.9 ± 0.6 | 17.9 ± 2.2* | 16.1 ± 2.8 |
| 28 | 8.8 ± 0.7 | 21.5 ± 2.8* | 17.9 ± 3.2# |
P < 0.05 vs. the corresponding value in the C group,
P < 0.05 vs. the corresponding value in the M group. Control: control group, MCT: monocrotaline group, PGE1: Prostaglandin E1 group.
Ratio of the medial thickening of pulmonary artery in each group (%).
Number of muscular pulmonary arterioles
The number of muscular pulmonary arterioles was significantly increased in the M group compared to that in the C group. From 21 to 28 days, the number of muscular pulmonary arterioles was significantly decreased in the PGE1 group compared to that in the M group (Table 3).
| Day | Control | M | PGE1 |
|---|---|---|---|
| 7 | 2.2 ± 0.1 | 2.3 ± 0.1 | 2.2 ± 0.4 |
| 14 | 2.2 ± 0.1 | 2.9 ± 0.1 | 2.6 ± 0.1 |
| 21 | 2.3 ± 0.2 | 3.8 ± 0.2* | 3.1 ± 0.2# |
| 28 | 2.4 ± 0.2 | 5.5 ± 0.5* | 4.2 ± 0.4# |
P < 0.05 vs. the corresponding value in the C group,
P < 0.05 vs. the corresponding value in the M group. Control: control group, M: monocrotaline group, PGE1: Prostaglandin E1 group.
Number of muscular pulmonary arterioles in each group.
Administration of PGE1 reduces RV and lung collagen deposition
To evaluate the effect of PGE1 treatment on the development of cardiac fibrosis, lung fibrosis, and remodeling, lung and RV samples were collected at 28 days after administration of MCT to determine histology patterns in the lung and RV. Results are shown in Fig. 1 and Fig. 2. After staining with Masson’s Trichrome, lung and RV sections showed more pronounced collagen deposition from animals with PAH compared to those in the control or PGE1 group (Fig. 1, P = 0.003 vs. C group, P = 0.01 vs. M group; Fig. 2, P = 0.003 vs. C group, P = 0.01 vs. M group).
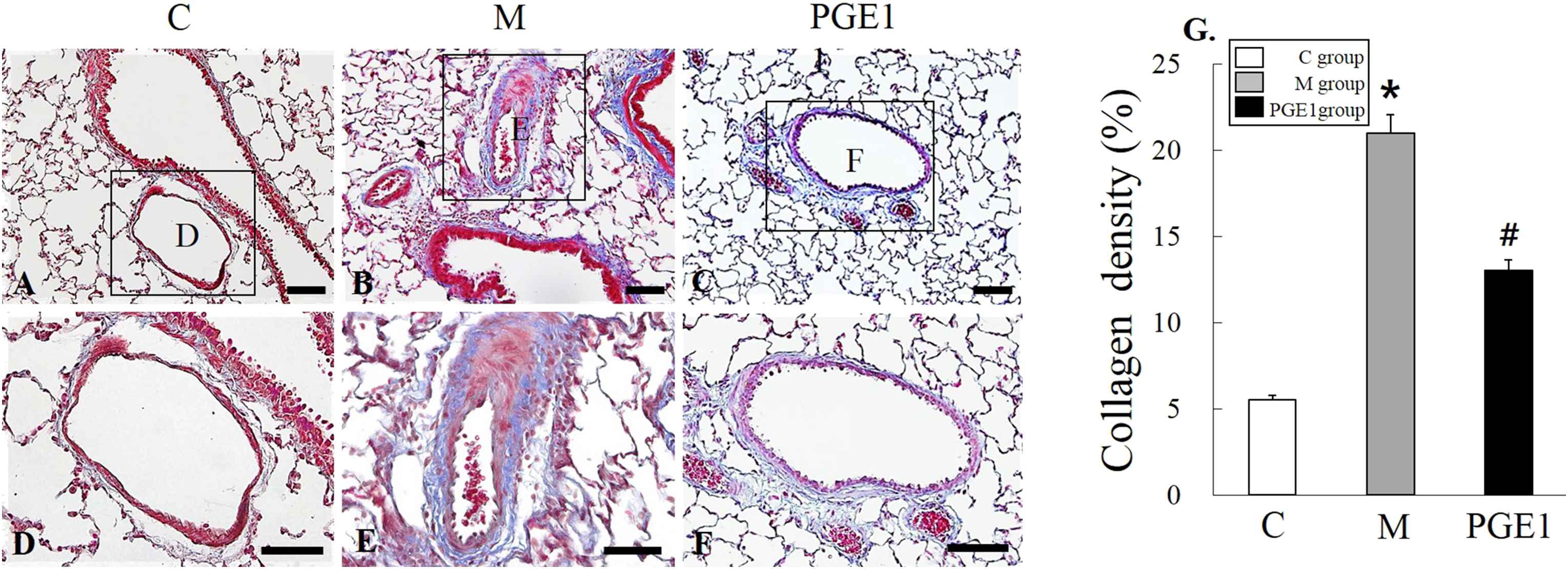
Representative images of lung stained with Masson’s Trichrome from control group (A), M group (B), and PGE1 group (C) at 28 days following the injection of MCT. Panels (D), (E) and (F) are high power views of panels (A), (B) and (C); fibrosis is colored blue. *P < 0.05 vs. C group, #P < 0.05 vs. M group. C group, control group; M group, monocrotaline group; PEG1 group, Prostaglandin E1. group. Scale bars = 150 μm (A, B, C), 50 μm (D, E, F). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
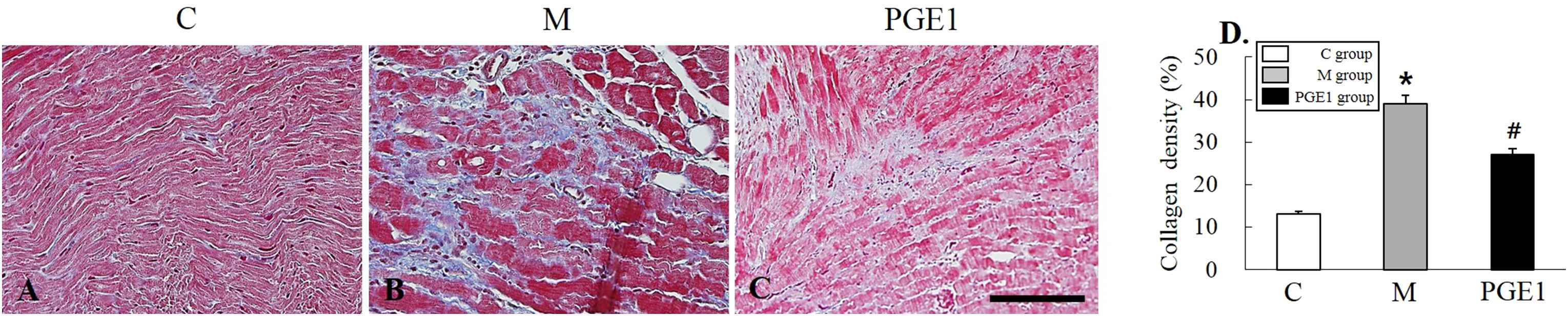
Representative images of right ventricle (RV) stained with Masson’s Trichrome from control group (A), M group (B), and PGE1 group (C) at 28 days following the injection of MCT. Panels (D), (E) and (F) are high power views of panels (A), (B) and (C); fibrosis is colored blue. *P < 0.05 vs. C group, #P < 0.05 vs. M group. C group, control group; M group, monocrotaline group; PEG1 group, Prostaglandin E1 group. Scale bars = 150 μm (A, B, C), 70 μm (D, E, F). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Immunohistochemistry analysis of lung samples
Immunohistochemistry (IHC) staining of lung tissues revealed that ET-1-positive cells were more prevalent in the PGE1 group, followed by those in the M group in comparison with those in the C group at 28 days (Fig. 3A–F). Three weeks after PGE1 treatment, ET-1-positive cells were still observed at the lung area in the PGE1 group. The increased level of ET-1 immunoreactivity observed in the M group was statistically significant (p < 0.05). The decreased level of ET-1-immunoreactivity was also significant in the PEG1 group compared to the M group (Fig. 3G).
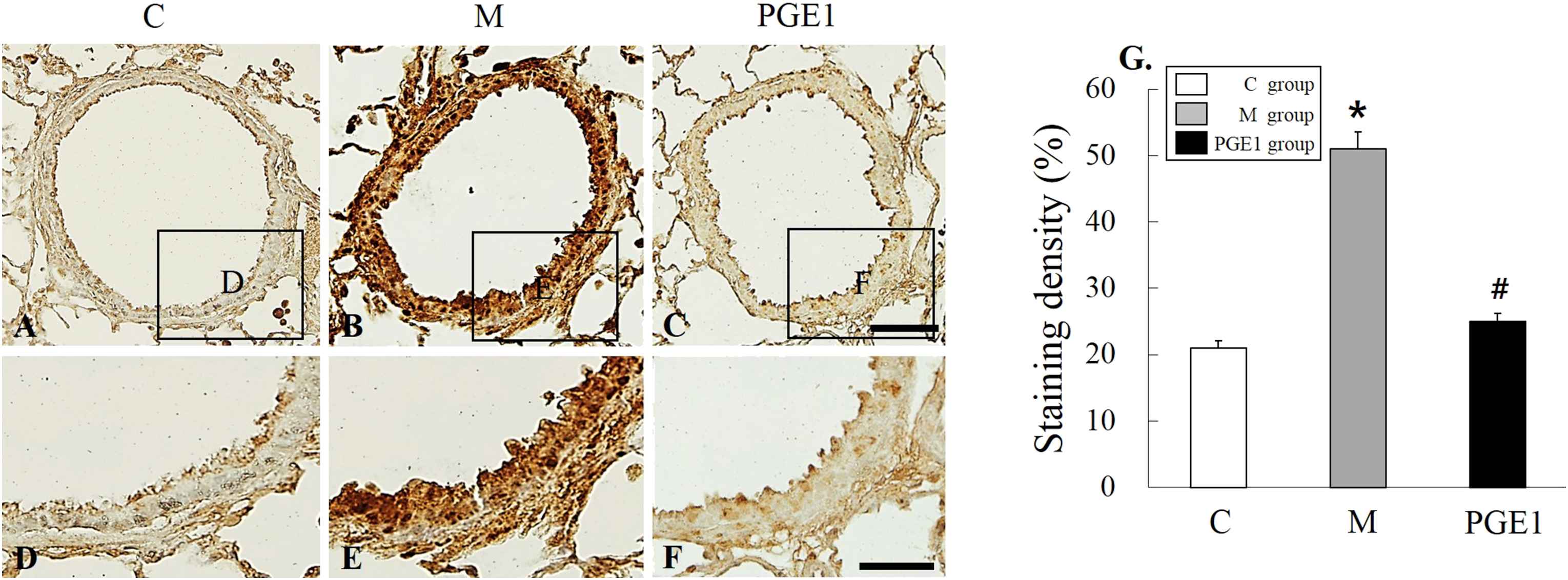
Localization of ET-1-immunoreactive cells in the lung tissues at 28 days (A–F). Immunohistochemical expression reveals that the positive cells of ET-1 is significantly higher in the M group than that in the C group, however it is lower in the PGE1 group than that in the M group. (G) Panels (D), (E) and (F) are high power views of panels (A), (B) and (C). C group, control group; M group, monocrotaline group; PEG1 group, Prostaglandin E1 group. *P < 0.05 vs. C group, #P < 0.05 vs. M group. Scale bars = 50 μm (A, B, C).
TUNEL apoptosis assay
TUNEL staining was performed to detect apoptotic DNA in lung tissues. In the C group, there was no positive staining (Fig. 4A and D). However, the M group had lung tissues with positive TUNEL staining (the presence of dark brown nuclei was observed) (Fig. 4B and E). PGE1 group also had cells with brown nuclei, indicating apoptotic DNA (Fig. 4C and F). Apoptotic cells were significantly more prevalent in the M group than those in the C group. However, they were less prevalent in the PGE1 group than those in the M group (Fig. 4G). These results indicated that PGE1 could attenuate apoptosis in the lung tissues of treated PAH rats.
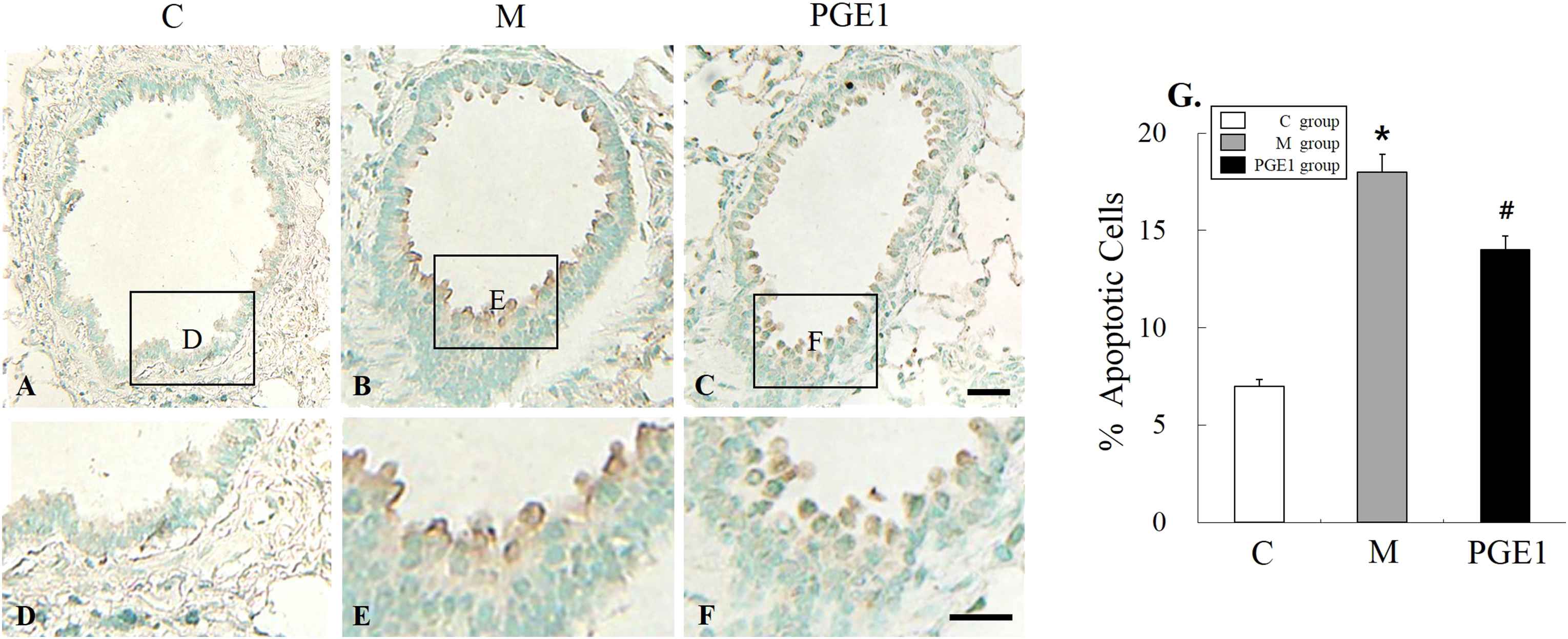
TUNEL assay on lung tissues at 28 days after PGE1 transfusion. Immunohistochemical expression revealed that the positive cells of apoptosis were significantly higher in the M group than that in the PGE1 group, however they were lower in the PGE1 group than that in the M group. *P < 0.05 compared with the C group. #P < 0.05 compared with the M group. C group, control group; M group, monocrotaline group; PEG1 group, Prostaglandin E1 group. Scale bars = 40 μm (A, B, C), 20 μm (D, E, F).
Western blot analysis
This experiment was designed to investigate if PGE1 group exhibited alterations in expression levels of ET-1 and ERA proteins in the lung. Changes in expression levels of ET-1 immunoreactivity in the lung were detected in the PGE1 group. Expression levels of ET-1 and ERA proteins detected in the PGE1 group were significantly decreased compared to those in the M group (Fig. 5A and B). Actin bands indicated protein loading in the same sample (Fig. 5, P < 0.0002 vs. C group, P = 0.0005 vs. M group).
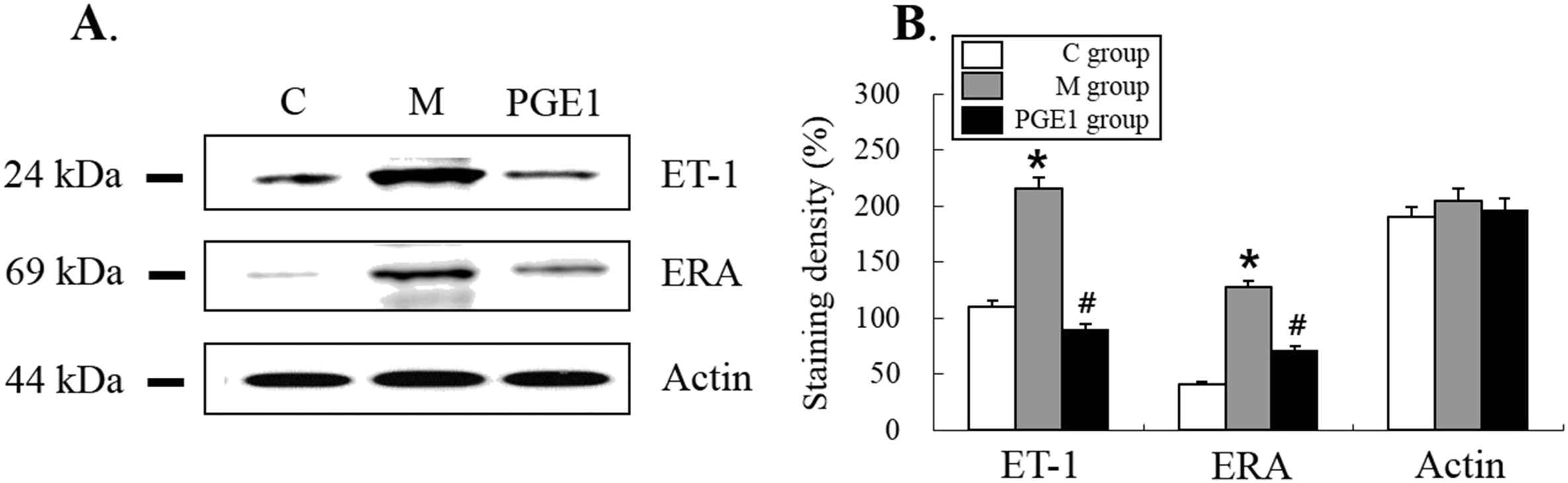
Western blot analysis of the levels of expression of ET-1, ERA and Actin immunoreactivity in the right ventricle (RV). The semi-quantitative analysis confirms that PGE1 group exhibited increased levels of immunoreactivity of ET-1 and ERA in the RV. C group, control group; M group, monocrotaline group; PEG1 group, Prostaglandin E1 group (n = 4–5 per group). *P < 0.05 vs. C group, #P < 0.05 vs. M group.
Discussion
The present study showed that treatment with PGE1 could reduce mal-adaptive structural remodeling of the lung and heart in MCT-induced PAH in rats. The main purpose of this study was to determine the effect of PGE1 on PAH using a rat model. Our results showed that PGE1 did reduce SPAP in PAH rats. It also improved RV hypertrophy, an indicator of RV pressure overload caused by elevated pulmonary artery pressure. In addition, it decreased the number of intra-acinar arteries and medial wall thickening in pulmonary arterioles, index of vascular remodeling.
The particular and beneficial effect of PGE1 could be due to its dual mechanism of action: one acute, predominantly vasodilatory effect on the pulmonary arterial system and another, more durable effect in chronic therapy, on the alveolar arterioles which depends on local mediators (endothelin, nitric oxide and angiotensin II).21 Indeed, these form the keystone to the neurohormonal approach in the treatment of heart failure. Furthermore, a direct action on muscle contractility should be mentioned. An AMP-dependent effect has been demonstrated on isolated atrial preparations by Scandinavian researchers,22 while in ischemic patients, a positive effect on plasma cytokine levels as interleukin-8, TNF-alpha was described.23 Previous studies have shown that endothelin-1 is a potent vasoconstrictor that plays a critical role in the pathogenesis of pulmonary hypertension.24–27 Reduction in pulmonary hypertension following captopril treatment was accompanied by a reduction in plasma endothelin-1.27 In the present study, there was a statistically significant reduction in endothelin-1 levels in the control and the PGE1 groups following the treatment (Figs. 3 and 5) However, a greater reduction of endothelin-1 was found in the PGE1 groups, suggesting that endothelin-1 reduction may have contributed to the improved pulmonary arterial hypertension in the PGE1-treated rats. Thus, there is compelling evidence showing that PGE1 is a pulmonary-selective vasodilator that can be used as an alternative to current therapy for PAH.
In the present study, we treated PAH rats with PGE1. We then performed H&E staining, Masson’s trichrome staining, immunohistochemical staining, and western blot analysis in order to investigate changes of ET-1 immunoreactivity in the cardiometer of these rats. A significant increase in pulmonary arterial pressure following MCT injection was accompanied by RV hypertrophy and increased lung and heart fibrosis when compared to the control group (Figs. 1 and 2). In the RV of rats with PAH, PGE1 treatment decreased the development of RV hypertrophy and normalized the expression of collagen content (Fig. 2). Additionally, in rats with PAH, SPAP of the heart was decreased by PGE1 treatment (Table 1). There results suggest that PGE1 could decrease pulmonary vessel contraction and remodeling, leading to improvement in apoptosis and inhibition of fibrosis in PAH rats. These findings support our hypothesis that chronic PGE1 administration can improve PAH.
PGE1 is a potent vasodilator in both pulmonary and systemic arteries.28 It has been used to treat postoperative PAH including PAH following mitral valve replacement or open-heart surgery in children.29–31 PGE1 infusion also improved ventricular function and survival in PAH secondary to advanced heart failure.32 One of the concerns regarding aerosol PGE1 administration in the past has been the potential incomplete absorption of the drug by the respiratory system, which may cause variations in therapeutic effects or side effects. In animal models of acute or chronic pulmonary hypertension, inhaled PGE1 increased ventricular contractility and reduced pulmonary arterial pressure.21,33 Inhalation of a single 25 μg dose of aerosolized iloprost, a prostanoid analogue, was associated with an enhanced cardiac index and reduced MPAP and pulmonary vascular resistance.34 The reasons for the greater pulmonary pressure reduction following PGE1 inhalation are not entirely clear. Therefore, the aim of this study was to evaluate the effect of continuous intravenous PGE1 on hemodynamics and long-term outcomes of rats with MCT-induced PAH. And, no study has attempted to develop a long acting PGE1. PGE1 might be used to treat cardiovascular disease. It has potential to induce vessel relaxation and inhibit excessive proliferation of vascular cells.
In conclusion, PGE1 can suppress PAH. Furthermore, PGE1 might be a new medicine for PAH. Further studies are needed to determine what pathways are involved in the improvement of PAH caused by PGE1.
Conflicts of interest
The author declares that he has no conflicts of interest to disclose.
Acknowledgement
The Korea Foundation for the Advancement of Science and Creativity (KOFAC) grant funded by the
References
Cite this article
TY - JOUR AU - Jae Chul Lee AU - Soo Young Choe AU - Chan Yeong Heo AU - Sun Ju Jeong PY - 2018 DA - 2018/05/26 TI - Prostaglandin E1 for preventing the progress of pulmonary arterial hypertension in rat model JO - Artery Research SP - 49 EP - 56 VL - 22 IS - C SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2018.05.001 DO - 10.1016/j.artres.2018.05.001 ID - Lee2018 ER -