
Sorafenib Induced Complete Cytogenetic and Molecular Response in a Chronic Eosinophilic Leukemia Case with t(12;13) Translocation
 , Serena Balducci1, *, †, , Francesca Guerrini1, Susanna Grassi1, Elena Ciabatti1, Claudia Baratè2, Maria Immacolata Ferreri3, Cecilia Giuliani3, Angelo Valetto3, Mario Petrini1, Sara Galimberti1
, Serena Balducci1, *, †, , Francesca Guerrini1, Susanna Grassi1, Elena Ciabatti1, Claudia Baratè2, Maria Immacolata Ferreri3, Cecilia Giuliani3, Angelo Valetto3, Mario Petrini1, Sara Galimberti1First co-author.
- DOI
- 10.2991/chi.k.200714.001How to use a DOI?
- Keywords
- Chronic eosinophilic leukemia; t(12;13); ETV6/FLT3; Sorafenib
- Copyright
- © 2020 International Academy for Clinical Hematology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
Hypereosinophilia (HE) is defined as a persistent increase in absolute eosinophil counts (AEC) to levels >1.5 × 109/L. HE can be ‘primary’ or ‘secondary’, more often accompanying infections, allergic disorders, autoimmune diseases or myeloproliferative/lymphoproliferative neoplasms [1,2]. In 2016, the WHO revised the classification of eosinophilic disorders, distinguishing myeloid/lymphoid neoplasms with eosinophilia and recurrent genetic lesions (such as rearrangement of PDGFRA, PDGFRB, FGFR1 and PCM1-JAK2), chronic eosinophilic leukemia (CEL) not otherwise specified (NOS), and idiopathic HE [3,4].
The ‘classical’ treatment of HE includes: (1) corticosteroids, which induce a significant reduction in AEC but are hampered by a frequent rapid rebound of these counts and of symptoms at tapering [4]; (2) hydroxyurea, which offers 70% of hematologic responses in steroid-refractory cases [5]; (3) interferon-alpha, successful in 50–75% of patients [4,6], even if accompanied by significant adverse events (transaminitis, cytopenia, mood alteration, hypothyroidism and “flu-like” symptoms). Variable hematological response rates have also been observed with cytoreductive agents, such as vincristine, cyclophosphamide, etoposide, 2-chlorodeoxyadenosine and cytarabine, but their use is limited to a few cases [4].
On the contrary, the modern approach to HE is a ‘patient-tailored’ strategy [7] based on identification of pathogenetic targetable molecular pathways: the FIP1L1–PDGFRA and PDGFRB rearrangements, for example, seem to be sensitive to the tyrosine kinase inhibitor imatinib [8]. For cases with FGFR1 rearrangement, a specific inhibitor, pemigatinib, is under investigation [4], while patients with a PCM1/JAK2 rearrangement can benefit from a JAK2-inhibitor, ruxolitinib [4].
Several cases of myeloproliferative neoplasms with eosinophilia showing non-recurrent translocations have been described, for which there are still no specific therapies. Among those, the t(12;13)(p13;q12) was first identified in 2006 by Vu et al. [9] in three patients showing myeloproliferative neoplasms with eosinophilia; the authors characterized the associated fusion gene as the ETV6/FLT3 rearrangement. FLT3 gene is located on chromosome 13q12.2 and codes for a class III tyrosine kinase receptor that controls proliferation and survival of hematopoietic progenitor cells [10]. The ETV6 gene (ETS variant gene 6) is located on chromosome 12p13.2; it encodes a nuclear transcription factor with a key role in hematopoiesis.
In the rare cases of ETV6/FLT3 fusion genes reported in literature, the ETV6 breakpoints were located in exons 4, 5 or 6, while FLT3 ruptures were found in exons 14 or 15 [11]. All reported cases with this fusion gene showed poor response and early relapse after conventional chemotherapy [11,12], but data concerning new drugs are still lacking. Indeed, FLT3 inhibitors, competitively inhibiting the ATP binding, thus blocking receptor autophosphorylation and activation of downstream signalling, could represent promising novel therapeutic compounds [13].
Here we present the case of a patient with CEL, NOS with ETV6/FLT3 rearrangement who achieved a complete hematologic, cytogenetic and molecular response after a few months of treatment with the FLT3-inhibitor, sorafenib.
On December 2017, a 69-year-old male, without significant comorbidities came to our attention after discovery of HE during routine exams. The blood cell count showed 21.97 × 109/L WBC, with markedly increased AEC (11 × 109/L), Hb 157 g/L and PLT 179 × 109/L. The patient complained of asthenia, weight loss and hyperpyrexia. The anamnesis and general tests excluded a secondary eosinophilia, so a bone marrow evaluation was performed. Morphological analysis showed an increased myeloblastic population comprising up to 10% of the total marrow cellularity and a markedly increased eosinophilic population. Cytogenetic analysis showed a translocation between the short arm of chromosome 12 and the long arm of chromosome 13, probably in p13 and q12, respectively, suggesting an ETV6/FLT3 rearrangement (Figure 1). To confirm this hypothesis, a qualitative semi-nested PCR was performed using ETV6-3F, ETV6-4F and FLT3-20R [9] primers, to allow the analysis of the N-terminal helix–loop–helix oligomerization domain of ETV6 gene and the juxtamembrane TK1 and part of TK2 domains of FLT3 [9]. As a positive control, the wild-type FLT3 was amplified with primers FLT3-14F and FLT3-20R. The PCR product, analysed by agarose gel electrophoresis, showed a visible band of about 300 bp (Figure 2). Subsequently, analysis by direct Sanger sequencing confirmed the suspicion of an ETV6/FLT3 rearrangement: the ETV6 exon 5 was fused to the FLT3 exon 19, preserving the reading frame from ETV6 through FLT3. Concurrently, internal tandem duplication (ITD) and tyrosine kinase domain (TDK) FLT3 mutations have been excluded using capillary electrophoresis and restriction enzyme digestions, respectively [14].
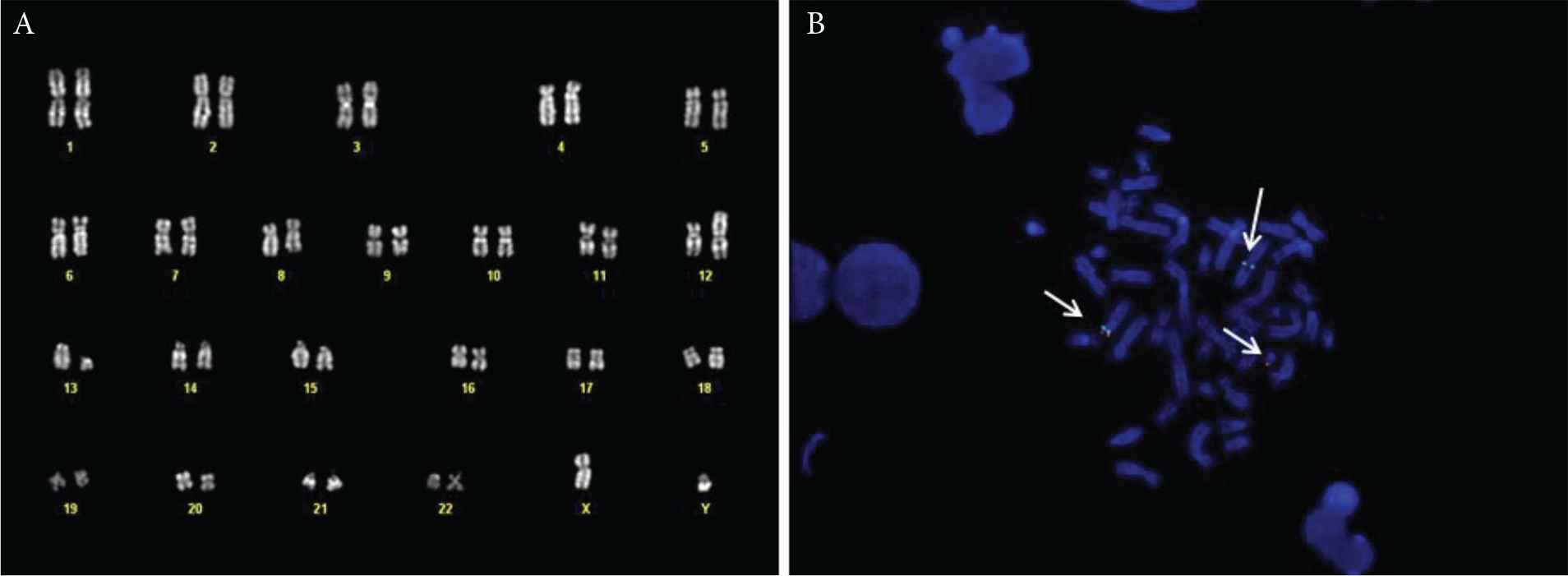
(A) Bone marrow karyotype of the patient, showing the t(12;13) translocation. (B) Fluorescent in situ hybridisation (FISH) with the ETV6 break-apart probe, showing the normal chromosome 12 (green and red signal), the derivative 12 (green signal) and the derivative 13 (red signal).
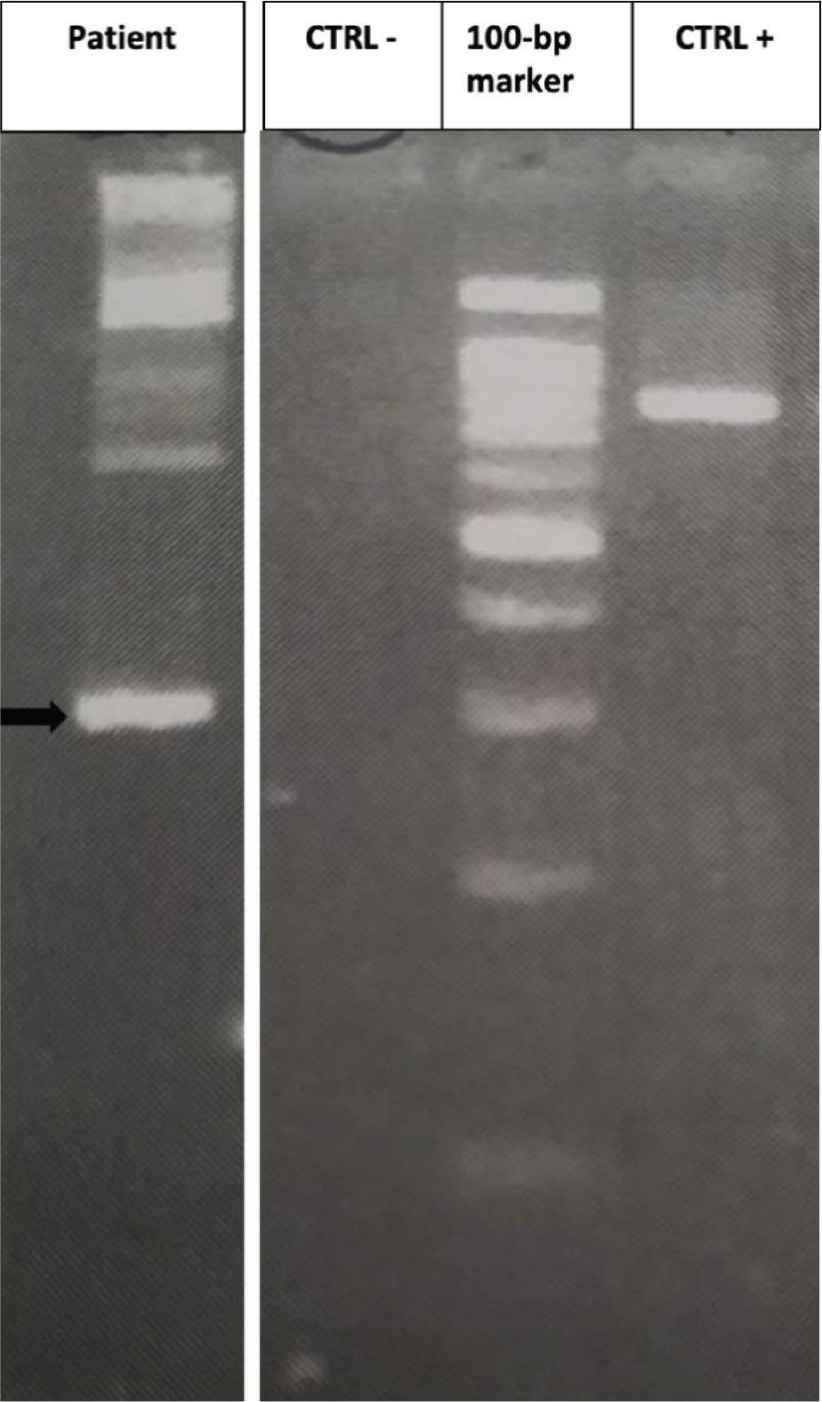
Results of semi-nested reverse transcriptase-polymerase chain reaction (RT-PCR). An ETV6/FLT3 band about 300 bp can be detected (black arrow); no band is present in the negative control (CTRL−); a band of about 800 bp can be seen when FLT3-14F/FLT3-20R are amplified (CTRL+).
As first line therapy, oral prednisone at 1 mg/kg/day was started to induce a rapid control of HE, without success. Thus, in February 2018, we decided to switch to a second line of treatment with interferon alpha, at an initial three MUI dose three times a week, which had to be rapidly reduced due to adverse events (severe flu-like syndrome and mood alteration). In May 2018, the blood cell count showed a partial hematologic response. In order to improve treatment efficacy and the patient’s quality of life, a pegylated formulation of interferon-alpha-2B was started at 30 μg/week. However, after 5 months of therapy the hematologic response was still not satisfying, with an AEC of 3.2 × 109/L. The bone marrow evaluation revealed a still increased eosinophilic population and only a modest reduction of the blast percentage, while cytogenetic analysis confirmed the persistence of t(12;13)(p13.1;3q12.11) in 50% of metaphases. The patient was considered ineligible for intensive chemotherapy. Therefore, after a review of the literature and acquisition of informed consent, we opted for an off-label use of the FLT3-inhibitor sorafenib. In April 2019 the patient started treatment at initial dosage of 400 mg/day. Unfortunately, he experienced multiple epistaxis episodes which imposed a dosage reduction to 200 mg/day. After 4 weeks of treatment, a complete hematologic response was obtained; in October 2019, after 6 months of therapy, the bone marrow assessment showed morphological, cytogenetic and molecular remission. However, weakness and mood alteration persisted; therefore, in views of the optimal response, we decided to further reduce the sorafenib dose to 100 mg/day.
Currently, after 12 months of therapy with the FLT3 inhibitor, the blood cell count confirms the complete hematologic response and the patient reports an excellent quality of life.
In summary, our experience supports the possibility of achieving a complete hematologic, cytogenetic and molecular response in patients with CEL NOS with ETV6/FLT3 fusion genes, thus emphasizing the importance of looking for any target lesions at diagnosis to set up an appropriate targeted therapy. For our patient, therapy with FLT3 inhibitors was crucial, even if it is still unknown whether this response will be long-lasting. In fact, some of the few cases previously reported in literature showed an acquired resistance due to the appearance of FLT3 mutations [15]. In our case, the breaking points involved in the formation of the ETV6–FLT3 rearrangement were on exon 5 of the ETV6 gene and on exon 19 of the FLT3 gene, therefore different from the breakpoints previously reported in literature. Whether this peculiar breakpoint may have determined a more indolent behaviour of CEL, remains to be ascertained. However, considering the excellent response, the fusion gene involving FLT3 was the right ‘target lesion’ in our patient.
Due to the reported short efficacy of FLT3 inhibitors, some studies are now investigating the use of different novel therapeutic strategies, such as heat shock protein (HSP) inhibitors [16]. Indeed, in a series of 75 patients with acute myeloid leukemia, it has been reported that FLT3-mutated cases presented high HSP levels, indicating a strong dependence of HSPs in stabilizing FLT3-encoded oncoproteins. Moreover, HSP90 inhibitors, in addition to their anti-proliferative effect, induce a reduction in pro-angiogenic chemokines levels, which may support the hypothesis of a long-term efficacy of these drugs [17].
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
FR, SB, CB and SG collected the data and wrote the paper. SG, FG and EC performed molecular biology analysis. MIF, CG and AV performed cytogenetic analysis. SG and MP revised the paper.
REFERENCES
Cite this article
TY - JOUR AU - Federica Ricci AU - Serena Balducci AU - Francesca Guerrini AU - Susanna Grassi AU - Elena Ciabatti AU - Claudia Baratè AU - Maria Immacolata Ferreri AU - Cecilia Giuliani AU - Angelo Valetto AU - Mario Petrini AU - Sara Galimberti PY - 2020 DA - 2020/07/27 TI - Sorafenib Induced Complete Cytogenetic and Molecular Response in a Chronic Eosinophilic Leukemia Case with t(12;13) Translocation JO - Clinical Hematology International SP - 129 EP - 131 VL - 2 IS - 3 SN - 2590-0048 UR - https://doi.org/10.2991/chi.k.200714.001 DO - 10.2991/chi.k.200714.001 ID - Ricci2020 ER -