
Idiopathic Aplastic Anemia: An Update
Peer review is under the responsibility of IACH
- DOI
- 10.2991/chi.d.190321.002How to use a DOI?
- Keywords
- Acquired aplastic anemia; Bone marrow failure
- Abstract
“Bone marrow failure” encompass all the conditions and syndromes in which there are qualitative or quantitative disorders of one or more lineages (erythroid, myelomonocytic, and/or megakaryocytic). A few years ago, the pathophysiology of these syndromes was completely unknown. Today we have better knowledge for these diseases, allowing the development of new treatment options and the improvement of patients' outcome. Acquired bone marrow failure syndromes include myelodysplastic syndromes, aplastic anemia, paroxysmal nocturnal hemoglobinuria, idiopathic neutropenia and large granular leukemia. All these syndromes share some common features and pathophysiology. The most important feature is the possibility of clonal evolution and progression into acute myelogenous leukemia, and open questions still remain on how to prevent evolution in these patients.
- Copyright
- © 2019 International Academy for Clinical Hematology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. BONE MARROW FAILURE
“Bone marrow failure” encompass all the conditions and syndromes in which there are qualitative or quantitative disorders of one or more lineages (erythroid, myelomonocytic, and/or megakaryocytic). A few years ago, the pathophysiology of these syndromes was completely unknown. Today we have better knowledge for these diseases, allowing the development of new treatment options and the improvement of patients' outcome.
Acquired bone marrow failure syndromes include myelodysplastic syndromes (MDS), aplastic anemia, paroxysmal nocturnal hemoglobinuria, idiopathic neutropenia, and large granular leukemia. All these syndromes share some common features and pathophysiology. The most important feature is the possibility of clonal evolution and progression into acute myelogenous leukemia, and open questions still remain on how to prevent evolution in these patients [1].
2. ACQUIRED APLASTIC ANEMIA
Acquired aplastic anemia [2] is a rare bone marrow failure syndrome characterized by pancytopenia in the peripheral blood and replacement of normal hematopoietic cells by fat in the bone marrow. Various factors are responsible for the development of aplastic anemia, such as drugs (i.e., chloraphenicol, nonsteroid anti-inflammatory drugs, chemotherapeutic agents), radiation, hepatitis viruses, pregnancy. In most cases where no etiologic factor can be found the diagnosis of idiopathic acquired aplastic anemia is made.
Idiopathic acquired aplastic anemia [3] is an autoimmune disease that affects mainly young people, usually around the second to third decades of life, with a small increase in presentation also noted in the fifth to sixth decades. Based on the peripheral blood counts, aplastic anemia can be moderate, severe or very severe. In all cases, the bone marrow cellularity is very low (below 30%). In very severe aplastic anemia, the absolute neutrophil count is below 200/μL, the platelet count is below 20,000/μL and the reticulocyte count less than 20,000. In severe aplastic anemia the absolute neutrophil count is below 500/μL, and in moderate aplastic anemia it is above 500/μL, but patients need platelet or red blood cell transfusions.
3. PATHOGENESIS OF ACQUIRED APLASTIC ANEMIA
3.1. T Cells
T cells have a central role in the pathogenesis of idiopathic acquired aplastic anemia [4]. Cytotoxic T cells are increased and constitutively activated, leading to high interferon-gamma production (Fig. 1). The T-bet transcription factor, which is responsible for the transcription of the interferon-gamma gene, is over expressed in T cells [5]. When patients respond to treatment, the T-bet l and interferon-gamma levels are is decreased. The antigen that stimulates and make T cells constitutively activated is not yet known; efforts have been made for auto antigens to be identified, without success up to date [6]. The regulatory T cell population is decreased in these patients, thus being unable to down regulate T cell activation [7]. Th17 cells are increased, and their numbers are inversely related to those of the regulatory T cells [8]. Increased interferon-gamma production is responsible for the Fas-mediated apoptosis and destruction of the hematopoietic stem cells in the bone marrow, leading to an empty bone marrow and pancytopenia in the peripheral blood. Additional alterations in T cells include low levels of perforin, SAP protein, and T cell receptor (TCR)-zeta chain, implicating defective constitutively activated T cells as the central player for stem cell destruction [9].
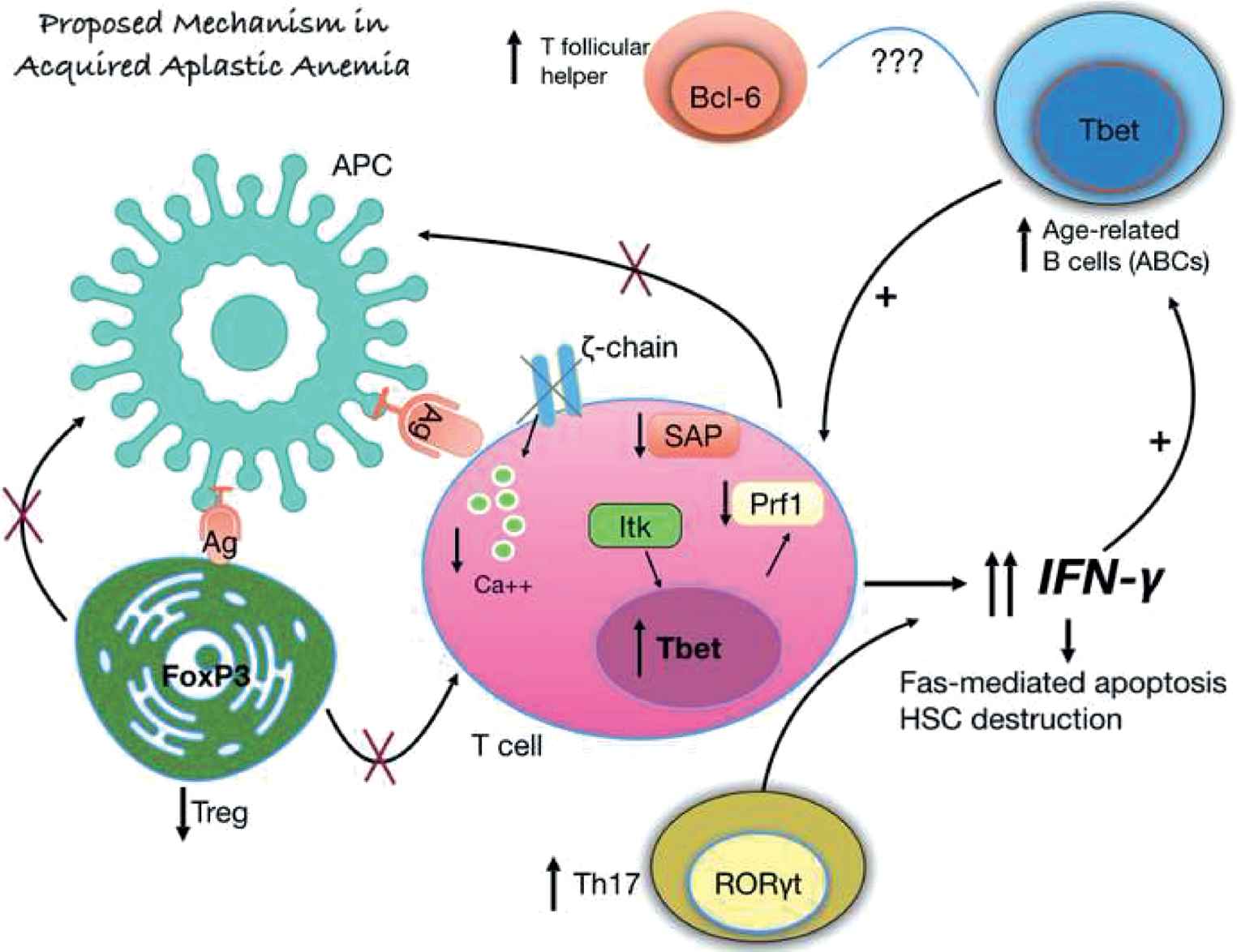
Proposed mechanism of interferon-gamma production and subsequent hematopoietic stem cell destruction in acquired aplastic anemia.
An unknown antigen is presented to T cells, leading to their activation. T cells in acquired aplastic anemia are constitutively activated with increased levels of the T-bet transcription factor, decreased perforin and SAP protein levels. Additionally, the zeta chain of the TCR is missing in some patients, and all these defects lead to an activated T cell that produces increased levels of interferon-gamma. The regulatory T cell population is decreased and the Th17 cells are increased. Additionally, the age-related B cells are also increased, further supporting interferon-gamma production and their relationship with the increased T follicular helper cell population (Solomou EE, unpublished data)
4. CYTOGENETIC ABNORMALITIES AND MUTATIONS
Until recently, it was thought that there were no cytogenetic abnormalities in patients with aplastic anemia. However, new molecular techniques revealed that approximately 20% of acquired aplastic anemia patients carry cytogenetic abnormalities or mutations [10]. The presence of clonal aberrations does not necessarily lead to evolution to MDS or acute myelogenous leukemia.
In spite of the difficulty in diagnosing aberrations with classical karyotypic analyses, due to the hypocellular bone marrow, approximately 12% of the patients will show abnormalities affecting most commonly chromosome 7 (usually monosomy 7), and also trisomy 8, 5q deletion, deletion 20q, deletion 13q, and trisomy 1. Trisomy 8 and deletion 13q herald a good prognosis in these patients [11]. Using single nucleotide polymorphism array (SNP-A) and fluorescence in situ hybridization (FISH) karyotypic abnormalities can be found in about 19% of the patients. Somatic mutations have been described in the TP53, ASXL1, DNMT3, and RUNX1 genes, are usually present in older patients, and are connected to worse prognosis. Mutations in the PIGA, BCOR, and BCORL1 genes are related to better prognosis. All studies so far agree that these mutations cannot predict which patients are going to evolve to myelodysplasia or leukemia, but only give information on overall survival and response to immunosuppressive treatment [12].
4.1. Telomeres
About 30% of the patients have shorter telomeres compared to healthy age-matched controls, but only 3–5% of the patients carry mutations in the ΤΕRΤ and TERC genes [13]. Telomeres are small repetitive hexamer DNA sequences, located at the edge of the chromosomes and protect from genetic material loss after each cell division. Studies have shown that acquired aplastic anemia patients with shorter telomeres have lower probability of responding to immunosuppressive treatment and a greater chance of relapsing if they respond [13]. Scheinberg et al. [14] study showed that patients can be grouped based on their telomere length and reticulocyte count at presentation: the group with shorter telomeres and lower reticulocyte counts had the worst outcome, were less likely to respond to immunosuppressive treatment, and exhibited low overall survival. On the other hand, patients with increased telomere length and more reticulocytes at presentation responded better and had the greater overall survival.
4.2. PIGA
Approximately half of the aplastic anemia patients will develop a Paroxysmal Nocturnal Hemoglobinuria (PNH) clone at some point during the disease course. The cells carrying the PIGA mutation are characterized by lack of Glycosylphosphatidylinositol (GPI)-linked proteins, making them more susceptible to complement lysis [15].
The presence of a PNH-clone usually results in better response to immunosuppressive treatment. This clone may represent a hematopoietic stem cell population that escaped from the cytotoxic T cells and has a survival advantage compared to the other hematopoietic stem cells. Flow cytometry analysis is the method of choice to detect even very small clones. Using PIGA sequencing, mutations in the PIGA gene have been identified, but only in the patients who had at least a 10% PNH clone by flow cytometry. In about 35% of the patients, double mutations in the PIGA gene have been identified. These mutations do not necessarily need next-generation sequence to be discovered. It is also known that PIGA gene mutations and small PNH clones are also present in healthy individuals; this is usually an age-related event. In contrast, in aplastic anemia the PNH clones and the PIGA mutations are not age-related.
4.3. Human Leukocyte Antigen System
In acquired aplastic anemia some Human Leukocyte Antigen System (HLA) (I and II) haplotypes, such as HLA-DR15, are over-represented [16,17]. These patients usually do better with immunosuppressive treatment. About 19% of aplastic anemia patients develop acquired chromosome 6p loss of heterozygosity (6pLOH). These 6pLOH cells possibly represent a population that escaped from the cytotoxic T cell destruction [18]. Also, these patients usually have specific haplotypes, mainly HLA-B*4002. They patients also respond better to immunosuppressive treatment, and rarely evolve to MDS. In contrast to the adult aplastic anemia population, the presence of HLA mutations in children is related to worse outcome after immunosuppressive treatment, more frequent detection of abnormal clones, and increased percentages of MDS-related mutations (ASXL1, DNMT3, and RUNX1).
4.4. BCOR/BCORL1
Patients with aplastic anemia have more frequent mutations in the BCOR/BCORL1 genes compared to those with MDS and healthy individuals [19]. These patients vary between 4% and 9% in different studies. None of the patients carrying the specific mutation evolve to MDS, and they show better response rates to immunosuppressive treatment and better overall survival. They usually also have PIGA gene mutations. The paradox is that patients with MDS who carry these mutations have worse prognosis and increased risk to develop acute myelogenous leukemia, while these clones usually remain stable or decrease over time in aplastic anemia.
4.5. TP53, ASXL1, DNMT3, and RUNX1
The clones carrying the TP53, ASXL1, DNMT3, or RUNX1 mutations usually increase over time. Despite this increase, these clones are smaller in aplastic anemia than in patients with MDS carrying the same clones [20]. The number of mutated genes is also higher in MDS patients as compared to aplastic anemia (mean mutated genes three compared to one in aplastic anemia). Patients with these mutations usually have worse prognosis and overall survival in comparison with those with mutations in the BCOR/BCORL1 and PIGA genes. Despite conferring a worse outcome, these mutations cannot predict who is going to evolve to MDS or acute leukemia.
4.6. Treatment Options
The age at disease diagnosis and the presence of a matched sibling donor remain the best factors to determine the treatment scheme [3] (Fig. 2). Younger patients, usually below the age of 40, with a matched sibling donor can immediately go into allogeneic bone marrow transplantation program, ideally less than 180 days after diagnosis. New studies revealed that patients less than 20 years old do better with transplantation [21]. All the treatment algorithms so far have the age of 40 as the cutoff limit for transplantation, but maybe this will change to the age of 20 years, due to differences in overall survival in patients with age below 20 and between 20 and 40 years [21]. The source of the graft should be bone marrow instead of peripheral blood as frequently used in other hematological diseases.
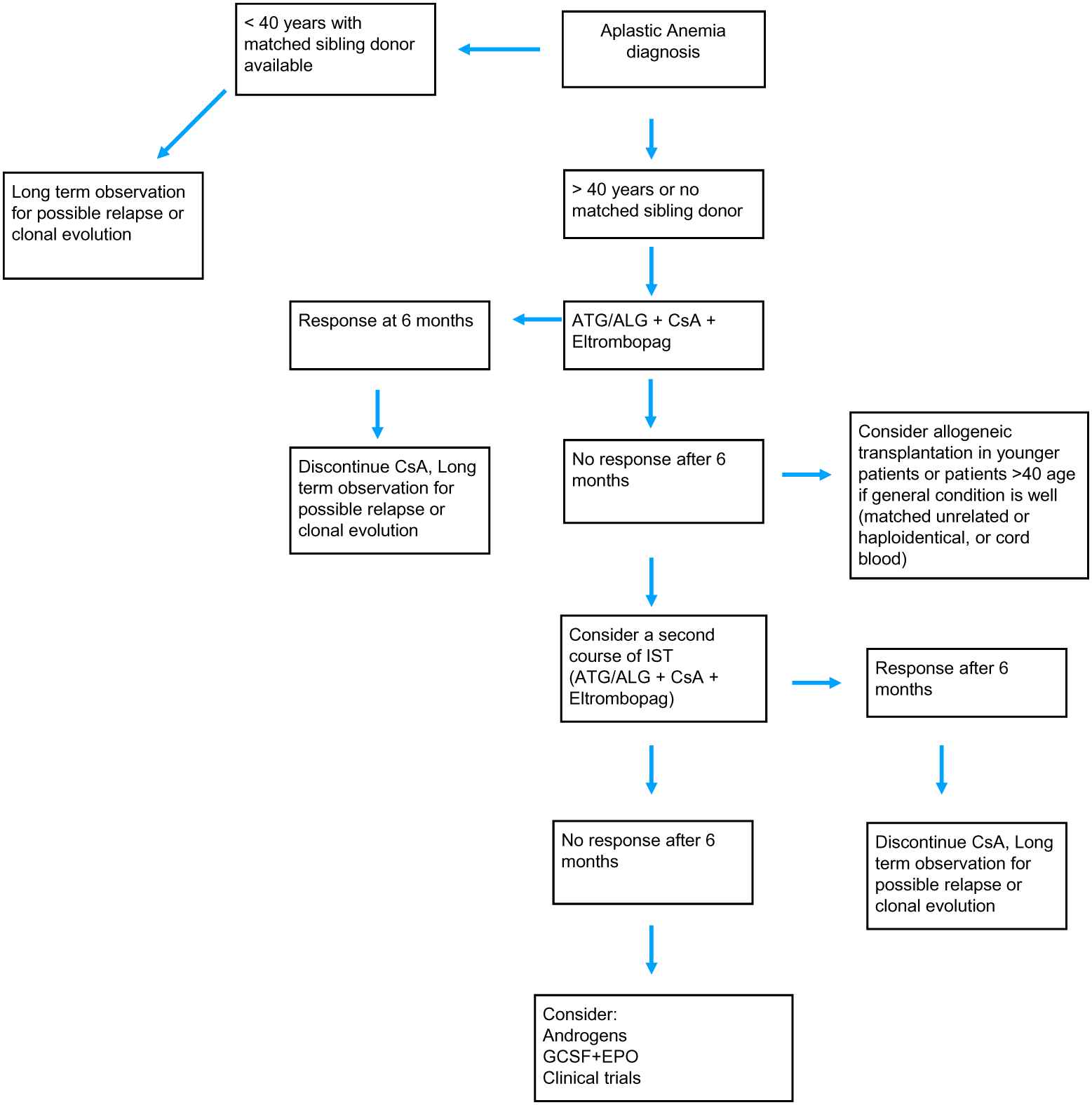
Treatment algorithm for the management of acquired aplastic anemia.
If the patient is older, or if there is no matched sibling donor available, the gold standard so far is immunosuppressive treatment with horse or rabbit anti-thymocyte globulins (ATG) plus cyclosporine. Very recently, eltrombopag was also approved to be given in newly diagnosed patients along with ATG plus cyclosporine as a first-line treatment option [22]. This triplet gives response rates ranging in different studies from 70% to 95% [23]. Eltrombopag binds to the trompopoietin receptor at a different position from that occupied by the endogenous thrombopoietin. Novel data showed that eltrombopag can overcome the inhibitory effect of the increased interferon-gamma levels on the thrombopoietin receptor, and this leads to the transcription of genes downstream the receptor pathway [24]. Eltrombopag is currently approved as a first-line treatment option in the United States, and the approval in Europe is expected for later this year. There are no data so far on the rate of relapse after the triplet combination. But the available data on the ATG plus cyclosporine treatment, which has been the gold standard for years, now show that approximately one-third of the patients will respond (with rabbit ATG) and one-third of these will relapse. A second course of immunosuppressive treatment can be given but, again, the response rate is not more than 30% [22].
Eltrombopag does not so far seem to increase the risk of clonal evolution, and is safe even in patients harboring mutations or karyotypic abnormalities. Patients with such alterations should be monitored regularly for additional mutations, karyotypic abnormalities, or clonal evolution [3].
There are ongoing trials in Europe and in the United States with the use of eltrombopag alone or in combination with cyclosporin for patients with moderate aplastic anemia. Trials with eltrombopag as the third drug in the triplet regimen (ATG + cyclosporin + eltrombopag) are also ongoing and should shed light on the long-term outcomes in severe aplastic anemia patients. These triplet regimen trials are very important because their results may change the practical approach in severe aplastic anemia established for many years. Other agents and approaches in ongoing trials include romiplostim, sirolimus, alemtuzumab, as well as the use of alternate donors (other than matched sibling donors) for bone marrow transplantation.
5. SUMMARY
In summary, acquired aplastic anemia is a rare autoimmune disease (Fig. 3). Cytotoxic T cells and increased interferon-gamma have a central role in the pathogenesis of the disease. With the new molecular techniques, mutations can be detected in approximately 50% of the patients, but they can only predict overall survival and response to immunosupressive treatment [25]. To date, no mutation profile can predict who is going to evolve to MDS or acute leukemia [26]. Eltrombopag is currently approved to be added to the so far gold standard immunosuppressive treatment with ATG and cyclosporine in newly diagnosed patients, with very promising response rates. Although a rare disease, aplastic anemia should be distinguished from other cases of pancytopenia, and patients should receive the best treatment available based on their age, and the existence of a matched sibling donor. The goal in the future is to determine the factors that lead to evolution into MDS or acute leukemia and prevent them.

Practical approach to a patient with acquired aplastic anemia.
REFERENCES
Cite this article
TY - JOUR AU - Elena E. Solomou PY - 2019 DA - 2019/04/01 TI - Idiopathic Aplastic Anemia: An Update JO - Clinical Hematology International SP - 52 EP - 57 VL - 1 IS - 1 SN - 2590-0048 UR - https://doi.org/10.2991/chi.d.190321.002 DO - 10.2991/chi.d.190321.002 ID - Solomou2019 ER -