
Endothelial nitric oxide synthase in the vascular wall: Mechanisms regulating its expression and enzymatic function
Authors equally contributed.
- DOI
- 10.1016/j.artres.2011.03.003How to use a DOI?
- Keywords
- eNOS; Nitric oxide; Tetrahydrobiopterin; Atherosclerosis
- Abstract
Endothelial nitric oxide synthase (eNOS) is the main source of nitric oxide (NO) in the vascular wall, a molecule with anti-inflammatory, antithrombotic, vasorelaxant, antioxidant and finally antiatherogenic properties. eNOS is expressed in vascular endothelium, and it uses
l -arginine as a substrate, while it also requires the presence of multiple co-factors such as tetrahydrobiopterin (BH4), nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) and others. In the presence of BH4 deficiency, this enzyme becomes uncoupled, and it is turned into a source of superoxide radicals instead of NO. Therefore, under these conditions which are present in patients with advanced atherosclerosis, eNOS in human vascular endothelium is largely a source of reactive oxygen species, inducing in this way atherogenesis. Therefore, the aim of future therapeutic strategies targeting atherosclerosis through regulation of eNOS physiology, should take into account that up-regulation of this enzyme in the vascular wall may not lead to a respective increase of NO bioavailability and improvement of vascular homeostasis, but it may actually induce intravascular oxidative stress, if intracellular bioavailability of eNOS co-factors is not simultaneously elevated. In conclusion, eNOS plays a critical role in the regulation of vascular homeostasis, and it is a therapeutic target against atherogenesis.- Copyright
- © 2011 Association for Research into Arterial Structure and Physiology. Published by Elsevier B.V. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Introduction
In 1980, Furchgott and Zawadzki1 demonstrated that the relaxation of vascular smooth muscle cells (VSMC) in response to acetylcholine is dependent on the anatomical integrity of the endothelium. The factor responsible for this intracellular relationship was named endothelium-derived relaxing factor (EDRF). In the late 1980s this factor was recognized as the free radical gas, nitric oxide (NO).
NO is synthesized by the enzyme NO synthase (NOS). The NOSs were first identified and described in 1989. NOSs catalyze NO biosynthesis through a reaction involving the conversion of
There are three distinct isoforms of NOS which differ both in structure and in function.4 Endothelial NOS (eNOS) and neuronal NOS (nNOS) are constitutively expressed and are referred to as Ca2+-dependent enzymes (even though eNOS can be activated in a Ca2+-independent manner).5 Inducible NOS (iNOS) is only expressed at high levels after induction by cytokines or other inflammatory agents, and its activity is independent of an increase of intracellular Ca2+.
The three NOS isoforms are characterized by regions of high homology (oxygenase and reductase domains) but at the same time each isoform exhibits distinctive features which reflect their specific in vivo functions.
The main source of endothelial NO is eNOS expressed by endothelial cells (ECs). Particular properties of eNOS which enable it to perform its specialized functions include Ca2+ sensitivity and the post-translational modifications which mediate sub-cellular localization. These enable the enzyme to respond not only to neurohormonal agents but also to hemodynamic forces. Although eNOS was often referred to as constitutive NOS, a number of factors such as hypoxia, estrogens and exercise are now known to alter its expression. Since endothelial control of vascular tone is a sensitive and highly tuned process, these changes are likely to be important in cardiovascular function especially in pathophysiological situations.
In addition to the well-studied role of NO in the process of penile erection,6 non-adrenergic, non-cholinergic relaxation occurs in all vascular smooth cells as a result of the expression of nNOS in peripheral neurons.7 In terms of enzymatic function nNOS appears to differ from other NOS isoforms by its readiness to catalyze the uncoupled oxidation of NADPH. Few are known for the reaction mechanism, even though it will help to understand the damaging role of nNOS in brain ischemia.8
iNOS expression can be induced by inflammatory mediators in most types of vascular cells, including ECs,9 cardiac myocytes,10 smooth muscle cells11 and macrophages.12 The expression of iNOS by macrophages and smooth muscle cells in atherosclerotic lesions has been taken as evidence for its determinant role in atherosclerosis.12 In addition iNOS expression is responsible for the impairment in eNOS-derived NO production in vessels treated with inflammatory mediators.13 On the other hand, iNOS expression may in some cases be protective, as shown by the iNOS-mediated suppression of allograft atherosclerosis, via the prevention of intimal hyperplasia.14
Endothelial nitric oxide synthase (eNOS) gene
Although several vasoactive factors are produced by the endothelium, the principal and best characterized is NO, produced by eNOS. The bioavailability of NO on the vessel wall is dependent on multiple factors such as the expression of eNOS, the presence of substrate and co-factors for eNOS, the phosphorylation status of eNOS, and the presence of reactive oxygen species (ROS) which can inactivate NO.
The three distinct genes for the human neuronal, inducible and endothelial NOS isoforms exist with a single copy each in the haploid human genome. The eNOS gene has 26 exons, 25 introns and it is located at 7q35–7q36 of chromosome 7. The protein consists of 1203 amino-acids and has a molecular weight of 133 kDA.15
Regulation of eNOS expression and NO availability
The ability of eNOS to produce NO is essential to vascular homeostasis. Disturbance of this ability is a major contributor to the pathogenesis of vascular disease. In vivo studies have shown that expression of eNOS is vital to endothelial function and have led to the understanding that eNOS expression is subject to significant degrees of regulation by numerous physiological and pathophysiological stimuli via mechanisms that alter steady-state eNOS mRNA levels. Such stimuli involve shear stress, hypercholesterolemia, estrogens, sphingosine 1-phospate, heat shock protein-90, transforming growth factor-β1, etc.
Laminar shear stress is one of the most potent regulators of eNOS mRNA expression. Laminar shear stress is a stimulus that has been shown to increase eNOS mRNA levels in vitro16 and in vivo.17 Transactivation of the eNOS promoter by laminar shear stress appears to be part of a negative feedback loop that involves NO.18 Laminar shear stress activates nuclear factor κB (NF-κB), and in addition, simultaneously increases enzymatic production of NO. The activation of NF-κB leads to translocation of p50/p65 heterodimers to the nucleus. The binding of these factors to the eNOS promoter leads to an increase in eNOS transcription, increasing eNOS protein levels, further augmenting NO production. The increased NO production ultimately leads to nitrophosphorylation of p50 and inhibition of NF-κB, resulting in termination of eNOS transcription.18 Dekker et al.19 were the first to demonstrate increased expression of lung Kruppel-like factor (KLF2) in human umbilical vein ECs (HUVECs) exposed to sustained shear stress. Recently it was established that this transcription factor is involved in laminar shear-induced eNOS transcription. These investigators showed that KLF2 mRNA is present in the endothelium of healthy human aortas, exclusively in areas of high flow. Subsequently, it was shown that adenoviral over expression of KLF2 in HUVECS resulted in a dramatic induction of eNOS mRNA, protein, and enzyme activity.20
Recently, it has been demonstrated that tumor necrosis factor α (TNF-α) also has effect on eNOS transcription. TNF-α decreased the activity of a human eNOS promoter construct that had been transiently transected into bovine aortic ECs, in a time- and dose-dependent manner.21 Although the TNF-α-dependent inhibition of promoter activity appeared to involve the NF-κB cascade, the loci mediating the inhibition were mapped to two Sp1-binding sites positioned between −109 and −95 and −81 and −67 relative to the transcription starting site.21 Interestingly, mutations of either loci greatly suppressed basal promoter activity, but only the upstream locus (−109 to −95) showed a decrease in Sp1/Sp3 binding activity in response to TNF-α.
Cyclosporine A (CsA) has been shown to paradoxically increase NO production both in vivo22 and in vitro23 considering the fact that therapy with this agent is associated with hypertension. Bovine aortic ECs exposed to CsA had a threefold increase in the expression of eNOS mRNA which was time dependent.22 Up-regulation of eNOS expression, induced by CsA has been proposed to be dependent on increased synthesis of ROS.24 Drummond et al.24 found that H2O2 increased eNOS mRNA levels up to fivefold in a time and dose-dependent manner.24 Analysis of the signaling cascade responsible for the effect of H2O2 on eNOS transcription established its dependence on Ca2+/calmodoulin-dependent protein kinase II (CaMKII) and janus kinase (JAK2).25 Through activation of CaMKII, H2O2, appears to be the mediator of increased eNOS expression that occurs in response to oscillatory shear stress.26
Lysophosphatidylocholine (lysoPC) has been shown to stimulate EC production of ROS27 and increases eNOS expression via enhanced transcription.28 Deletion analysis of human eNOS promoter-luciferase construct has identified Sp1 site at −104 to −90 and PEA3 sites at −40 to −24 as being involved in lysoPC-induced promoter activity.29 Gel-shift assays revealed that lysoPC augmented Sp1 binding activity. Subsequent analysis of the signaling events involved in the stimulatory effects of lysoPC on promoter activity, revealed a PI-3Kγ-related pathway.30 In this pathway PI-3Kγ activated JAK2, which in turn activated meiosis specific kinase 1 (MEK1). MEK1 stimulated Sp1 binding to the eNOS promoter through activation of Extracellular Signal-Regulated Kinases 1 and 2 (ERK1/2) and subsequently increased protein phosphatase 2A activity.29,30 ERK1/2 exerted its effect through reducing the control of protein phosphatase 2A by casein kinase 2.31
In both cell culture and animal studies, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (HMG-CoA reductase; statins) have been shown to increase eNOS expression.32 In human saphenous vein ECs, simvastatin and lovastatin both increased eNOS mRNA and protein levels in a time- and dose-dependent manner.32 The mechanism responsible for this effect was found to be post-transcriptional; simvastatin treatment did not alter the rate of eNOS transcription but significantly prolonged eNOS mRNA half-life as assessed by actinomycin D transcriptional arrest studies. In addition, these studies showed that simvastatin and lovastatin both counteracted the downregulation of eNOS expression by hypoxia and oxLDL,32 two stimuli known to decrease eNOS mRNA stability.33 By inhibiting HMG-CoA reductase, statins prevent the synthesis of isoprenoid intermediates in the cholesterol biosynthetic pathway.33 These intermediates serve as important lipid attachments for the post-translational modification and activity of a variety of signaling proteins. These proteins are members of the Rho GTPase family.34 Laufs and Liao35 found that Rho negatively regulates eNOS expression in human ECs; Rho inhibition increased eNOS expression, and Rho activation diminished eNOS expression. Furthermore, treatment of ECs with statins decreased the geranylgeranylation, membrane translocation, and GTP binding activity of Rho. Therefore, statins appear to upregulate eNOS expression by blocking Rho geranylgeranylation. Interestingly, geranylgeranyl modification also appears to be involved in trichostatin A (TSA)-induced downregulation of eNOS expression. However, this latter mechanism is not dependent on Rho signaling.36
TGF-β1 is a 25-kDa peptide that plays an important role in the pathogenesis of atherosclerosis, hypertension and angiogenesis.36 Treatment of bovine aortic ECs with TGF-β1, increased eNOS mRNA in a time- and dose-dependent manner.37 TGF-β1 increases eNOS transcription via recruitment of multiple transcription factors such as Smad2 and NF-1 to distinct cis-acting sequences.37
Heat shock protein 90 (hsp90), modulates agonist-dependent eNOS activation.38 Hsp90 binding stimulates eNOS activity, and also affects eNOS specific activity by binding to kinase Akt. This interaction may be dynamically regulated by hsp90 S-nitrosylation.39
Sphingosine 1-phosphate (SP1) and lyosphosphatidic acid activate endothelial G-protein-coupled S1P receptors (formerly called EDG receptors) and play key roles in vascular regulation40 by stimulating AMP-activated protein kinase and the small GTP binding protein Rac1, critical determinants of the activity of eNOS.
The potentiating effect of estrogens on endothelium-dependent relaxations involves both genomic and non-genomic effects.41,42 Phytoestrogens and selective estrogen receptor modulators also potentiate endothelium-dependent relaxations/vasodilatations.42
High levels of cholesterol in humans affect endothelium-dependent relaxations/dilatations and the normalization of the cholesterol level with treatment restores endothelial function.43 The effect of hypercholesterolemia is due to the combination of increased oxidative stress leading to a reduced bioavailability of NO, impairment of eNOS activity, and augmented levels of circulating asymmetric dimethylarginine (ADMA)42 (Table 1).
| Positive Regulation | Negative Regulation | ||
|---|---|---|---|
| Activity | Expression | Activity | Expression |
| Laminar shear stress20 | Laminar shear stress16 | Hypercholesterolemia43 | TNF-α21 |
| S1P40 | LysoPC28 | Ox-LDL33 | Hypoxia33 |
| Hsp9038 | VEGF143 | ADMA88 | |
| Estrogens41 | Caveolin57 | ||
| Sphingolipids40 | ROS144 | ||
| Bradykinin145 | Ang II92 | ||
| Adiponectin146 | |||
S1P: Sphingosine-1-phosphate, Hsp90: Heat Shock Protein 90, LysoPC: Lysophosphatidylocholine, VEGF: vascular endothelial growth factor, TNF-α: Tumor necrosis factor α, Ox-LDL: Oxidized-LDL, ROS: Reactive oxygen species, ADMA: Asymmetric dimethylarginine, Ang II: Angiotensin II.
Factors affecting eNOS activity and expression.
Common genetic polymorphisms of eNOS gene
To date more than 100 polymorphisms have been identified in, or in the vicinity of, the gene. More than 15 polymorphisms exist in the eNOS promoter that might influence mRNA transcription and reduce gene expression.
The −786T/C promoter polymorphism influenced transcriptional activity in vitro in a luciferase/reporter assay system and was associated with coronary arterial spasm in Japanese subjects.44 In one study, ECs from subjects with the CC genotype exhibited reduced shear stress induced eNOS mRNA transcription, and vascular rings from such subjects had diminished endothelium-dependent vasodilatation.45
Polymorphisms within the coding region of the eNOS gene could alter NOS enzymatic activity. One of the most studied NOS polymorphisms (G894T) within exon 7 is the only common polymorphism identified until now, that encodes an amino acid substitution-Glu298Asp (glutamate to aspartate position 298).46 Two studies have shown that eNOS Asp298 is subjected to selective proteolytic cleavage in ECs and vascular tissues, that might account for reduced vascular NO generation.47 Data from physiological studies have also been inconsistent. In some studies, individuals with the Asp298 allele exhibited altered vascular responses, for example a reduced blood pressure fall following exercise training, a lower basal blood flow and reduced vasodilatation to adenosine in the coronary arteries, an enhanced systemic pressure response to phenylephrine, a reduced flow mediated dilatation (FMD) of the brachial artery if concurrent smokers, impaired dilator responses to acetylcholine if hypertensive, or impaired enhancement of FMD during the first trimester of pregnancy.46 Moreover, we have demonstrated that this polymorphism affects endothelial function in patients with atherosclerosis,48 while it also plays a role in the regulation of systemic low-grade inflammation during myocardial infarction, an effect which is however disappeared in these patients under resting conditions, one year post-infarction.49,50 The impact of this polymorphism on cardiovascular risk was also demonstrated in a large meta-analysis by Casas et al.51 Despite the numerous data from clinical studies, some experimental data have questioned the association of this genotype with functional phenotype of eNOS.52
eNOS molecular structure
eNOS exhibits a bidomain structure, in which an N-terminal oxygenase domain containing binding sites for haem, BH4 and
There are three distinct isoforms of NOS which are different both in their structure and function. Endothelial NOS (eNOS, NOS III, 2 × 134 kDa) and neuronal NOS (nNOS, NOS I, 2 × 160 kDa) are referred to as constitutively expressed, Ca2+-dependent enzymes although eNOS can be activated in a Ca2+-independent manner.5 iNOS (NOS II, 2 × 130 kDa) is expressed at high levels only after induction by several inflammatory agents, and its activity is independent of Ca2+ increase.
Of the three NOS isoforms, eNOS alone is acylated by both myristate and palmitate.53 eNOS is co-translationally and irreversibly myristoylated at an N-terminal glycine residue while palmitoylation occurs post-translationally and reversibly at cysteine residues Cys15 and Cys26.54 Dual acylation of eNOS is required for efficient localization to the plasmalemmal caveolae of ECs.55 Palmitoylation is dynamically regulated by agonist (e.g. bradykinin)-induced changes in intracellular Ca2+.56 eNOS is localized to the caveolae,55 which are microdomains of the plasmalemmal membrane that are implicated in a variety of cellular functions including signal transduction events. The caveolin proteins are the major coat proteins of caveolae and in ECs eNOS binds to caveolin-1, while in cardiac myocytes eNOS is associated with caveolin-3.57,58 Caveolin 1 and peptides from the caveolin-1 scaffold region directly inhibit eNOS activity and this interaction is regulated by Ca2+/CaM.57,58 A nine-amino-acid binding motif for caveolin has been identified in bovine eNOS, residues and deletion mutation of these amino-acids produced an active enzyme that was uninhibitable by caveolin-1.59 Additionally, an independent interaction between caveolin-1 and the reductase domain of eNOS has been described.60
eNOS enzymatic function
Biosynthesis of NO involves a two-step oxidation of
Even though the reductase and oxygenase domains are able to function independently under certain circumstances, NO synthase activity is carried out by the homodimer.
Haem plays an essential role in the dimerization of eNOS. In its absence, eNOS, and all the other isoforms exist in monomers, which are unable to bind BH4 or a substrate analog and do not catalyze the
eNOS coupling and the role of vascular tetrahydrobiopterin regulation
The NOS cofactor BH4 is essential for the proper transport of electrons to
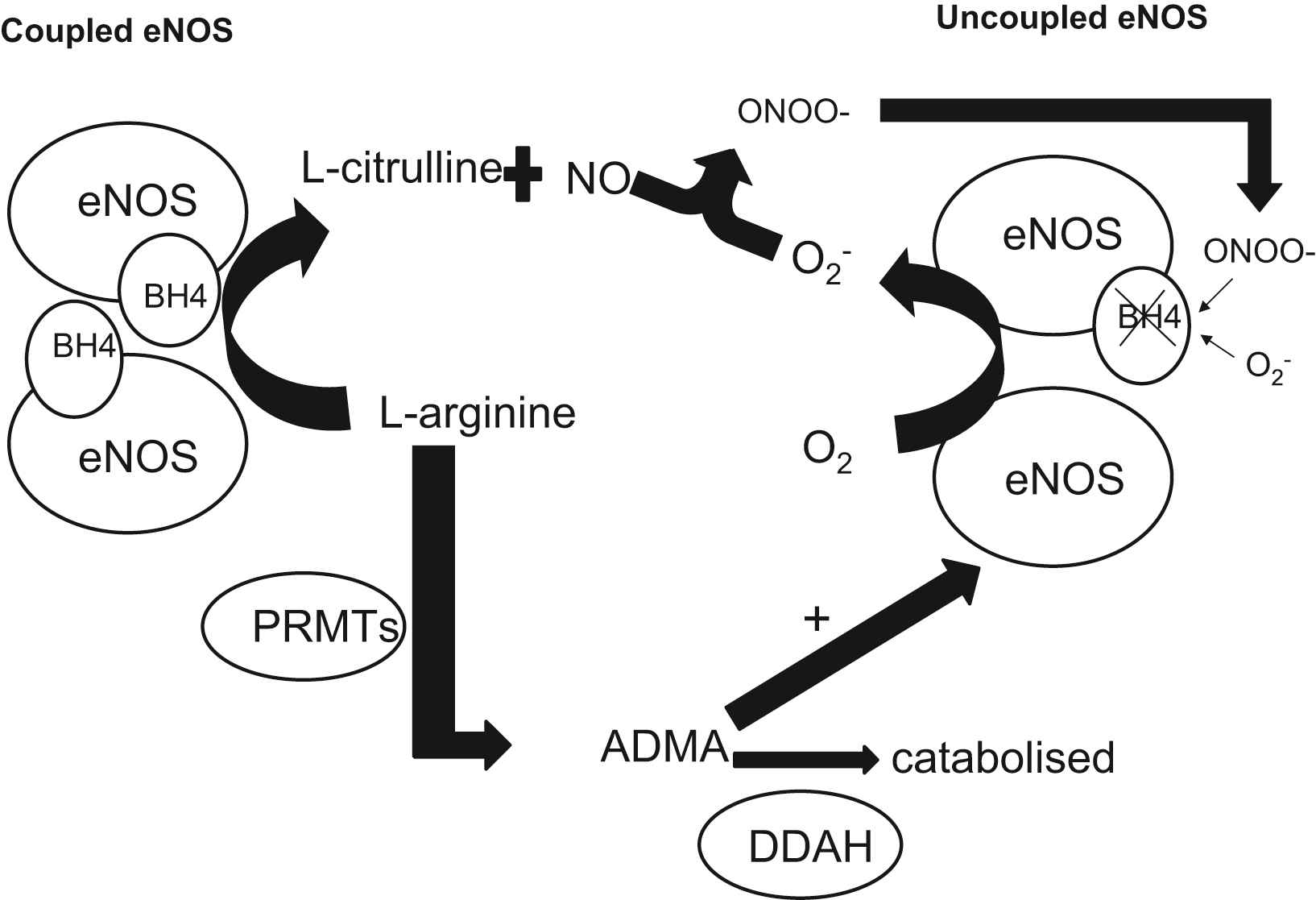
The role of tetrahydrobiopterin (BH4) and asymmetrical dimethylarginine (ADMA) on endothelial nitric oxide synthase (eNOS) coupling: The NOS cofactor BH4 is essential for the proper transport of electrons to
De novo production of BH4 is reliant upon three enzymes: GTP cyclohydrolase I (GTPCH), 1,6-pyruvoyl-tetrahydropterin synthase, and sepiapterin reductase.71 The initial step in BH4 synthesis is the conversion of GTP to dihydro-neopterin triphosphate by GTPCH, which is the rate-limiting enzyme in this biosynthetic pathway and is encoded by the GCH1 gene.72 This gene is characterized by two different haplotypes, termed X and O, which are defined by 3 polymorphisms: rs8007267G_A, rs3783641A_T, and rs10483639C_G (X haplotype: A, T, G; O haplotype: any other combination).73 The presence of the X haplotype in patients with multivessel CAD has been associated with lower levels of both plasma BH4 and total biopterins (which is the sum of BH4, BH2, and B, reflecting the overall biosynthetic activity of GTPCH), as well as significantly lowers vascular GCH1 mRNA expression and vascular BH4 levels.74 This effect was not only observed in the XX genotype but also in the XO genotype, suggesting that the X haplotype may be an important and common factor in regulating circulating biopterin levels in patients with CAD.
Several in vitro studies suggested that GCH1 expression in human macrophages and endothelial cells is induced by proinflammatory cytokines, resulting in an elevation of intracellular BH4 in endothelial cells.75,76 However, this effect was only observed after simultaneous exposure to high concentrations of multiple cytokines, which is not as likely to occur in a clinical setting. In contrast, we have shown that in patients with CAD, systemic inflammatory stimuli that can cause an elevation in plasma biopterins lead to a decrease in vascular biopterins, which are mainly located in the endothelium.77 Plasma biopterins are positively correlated with CRP levels and inversely associated with endothelial function, whereas vascular BH4 was associated with maintained eNOS coupling, reduced vascular O2− and improved endothelial function. These findings delineate a discordant regulation of circulating versus vascular biopterins, thus linking systemic inflammation with endothelial dysfunction and oxidative stress.
Oxidative inactivation of BH4 probably occurs through reaction with ROSs generated within the vessel wall, mainly peroxynitrite (ONOO−) and O2−.77 The resulting uncoupling of eNOS leads to generation of O2− which react with NO to form ONOO-, further oxidizing BH4 in a feed-forward mechanism. Local intravascular administration of BH4 to smokers, hypertensives or hypercholesterolemics can reinstate normal endothelial function, whereas BH4 has no effect on healthy vessels at the same dose.70 This observation suggests that BH4 availability may be limited in diseased vessels. In addition, we have previously reported that intravenous administration of 5-methyl-tetrahydrofolate (5-MTHF), the circulating form of folic acid, lead to an amelioration of eNOS function, improving NO bioavailability and reducing vascular oxidative stress in arteries and veins of CAD patients, both in vivo and ex vivo.78 Our findings suggest that although 5-MTHF is a rather weak superoxide scavenger, it may act by preventing ONOO−-induced oxidation of BH4 in the vascular wall and increasing intracellular BH4 bioavailability. Similarly, oral folate supplementation improved endothelial function in CAD patients, partly through improved BH4 bioavailability.79 This beneficial effect was similar between patients receiving high and low-dose supplementation, suggesting that it is mediated through an increase in vascular 5-MTHF levels rather than plasma 5-MTHF levels, which are dose-dependent.80 In a very recent study, we demonstrated that the functional C677T polymorphism in the methyl tetrahydrofolate reductase (MTHFR) gene, which is associated with lower plasma and vascular 5-MTHF levels, exerts a direct effect on vascular BH4 levels, eNOS coupling and NO bioavailability in human vessels in vivo.81 These results propose that physiological variations of vascular and plasma 5-MTHF levels could reflect endothelial function in humans. In addition, Lemarie et al.82 showed that MTHFR deficiency was associated with increased ROS production and reduced NO generation in endothelial progenitor cells (EPCs), heterozygous for the gene deletion MTHFR (+/−). Furthermore, treatment of EPCs with sepiapterin, a precursor BH4 significantly reduced ROS and improved NO production.
ADMA is a naturally occurring amino acid that circulates in plasma, is excreted in urine, and is found in tissues and cells.83,84 ADMA is synthesized when arginine residues in proteins are methylated by the protein arginine methyltransferases (PRMTs).85 The increase in ADMA levels reflects the expression and the activation status of the dimethylarginine dimethylaminohydrolases (DDAH-1 and DDAH-2). The significance of this pathway is highlighted by the fact that the transgenic over expression of DDAH-1 has been shown to increase NO production and reduce blood pressure in vivo86 while its deletion is associated with endothelial dysfunction and high blood pressure.87 Interestingly, it seems that although the downregulation of both DDAH enzymes can affect endothelial NO production, only the loss of DDAH-1 was associated with a reduction in the arginine/ADMA ratio88 indicating that DDAH-2 may affect NO output by an ADMA-independent mechanism. We have demonstrated that elevated circulating ADMA is associated with increased vascular O2− generation by uncoupled NOS in patients with CAD.89 In this population however, vascular eNOS is largely uncoupled as a result of the decreased bioavailability of BH4 in the vascular wall77 and until recently, the interaction between ADMA and uncoupled eNOS was largely unidentified. In experiments with purified eNOS, it was demonstrated that ADMA increases uncoupled NOS-derived O2− generation, only in the presence of BH4 deficiency.90 This has been suggested to be due to the ADMA-induced elevation of NADPH and O2 consumption by the uncoupled enzyme.
Therapeutic strategies targeting NO availability
The understanding of eNOS molecular biology as well as the mechanisms that regulate its expression and activity has turned the therapeutic targenting of several vascular diseases such as atherosclerosis and coronary artery disease in this direction (Table 2).
| Therapeutic agent | Comment/selected most important mechanisms |
|---|---|
| ACEi/AT1R blockers | Improvement of intracellular redox state, preventing oxidative degradation of co-factors.93 |
| Increase of eNOS activity by increasing eNOS phosphorylation at Ser1177 and bradykinin levels.94 | |
| Increase of eNOS expression by suppressing TNF-α activity.99 | |
| Statins | Reduction of overall ROS pool, preventing the oxidation of co-actors and improving coupling.105 |
| Increase eNOS expression, by stabilizing mRNA and reducing Nuclear factor kappaB and other transcriptional pathways.106 | |
| Increase eNOS activity by modifying P13-kinase – Akt protein kinase pathway.106 | |
| TZDs | Improve eNOS coupling by affecting eNOS phosphorylation at Ser1177.114 |
| Modify heat shock protein 60-eNOS interaction, affecting eNOS activity.114 | |
| Reduce gp91phox expression and modify NADPH-oxidase activity; by reducing overall cellular oxidative stress status improve eNOS coupling/activity and stimulate its expression.116 | |
| Folates | Reduce homocysteine, therefore exert intracellular antioxidant properties; stimulate eNOS expression.80 |
| Scavenge peroxynitrite (and less superoxide) directly, therefore protecting the oxidation of BH4 and improving eNOS coupling.78 | |
| BH4 | Improve eNOS coupling and activity.147 |
| Vitamins C and E | Antioxidant properties. Improves the activity/coupling of eNOS by improving intracellular BH4 bioavailability.125 |
| Polyphenolic antioxidants | Antioxidant properties. Reduce NADPH oxidase activity, increase eNOS activity.126–128 |
| ET-antagonists | Endothelin ETA receptor blockade. Increases NO/ |
| Aliskerin | eNOS activity, reduces O2− and ONOO−.102 |
ACEi: angiotensin-converting enzyme inhibitor, AT1R: angiotensin II receptor, TZDs: Thiazolidinediones, ROS: Reactive oxygen species, ADMA: Asymmetric dimethylarginine, Hcy: Homocysteine, BH4: Tetrahydrobiopterin, ET: endothelin.
Therapeutic strategies targeting eNOS expression and function.
Angiotensin converting enzyme inhibitors and angiotensin receptor blockade
As it is well-known angiotensin II (Ang II) exerts pro-oxidant, proinflammatory and proliferative effects on the vasculature mainly via the constitutively expressed angiotensin II type 1 receptor (AT1R).91
Angiotensin converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARBs) exert their effect on endothelial function partly via the increase of NO bioavailability. Further to its well known atherogenic and proinflammatory properties, Ang II contributes to the ROS pool by activating NADPH oxidase activity resulting to the reduction of NO bioavalablity.92 One of the mechanisms by which ACEi exert their effect on NO availability increase is the reduction of ROS generation. As it was demonstrated by Lob et al. ACEi reduced the Ang II stimulated intracellular production of ROS.93 Another mechanism, which probably is responsible for the effect of ACE inhibition on NO availability is the prevention of bradykinin breakdown, thereby increasing kinin-dependent nitric oxide release mediated by the B2-kinin receptor. Several animal studies demonstrated that the effects of ACEi on NO release are blocked by B2-kinin receptor antagonists.94,95
Blockade of the angiotensin II AT1 receptor reduces the vascular signaling and the production of ROS that suppress nitric oxide activity. As Taddei et al96 demonstrated, candesartan inhibited the vascular production of ROS in ApoE-deficient mice. In addition, the synergic use of an ACEi metabolite and an ARB resulted in an increase of bradykinin and cGMP in sodium depleted rats.97 Moreover, a five-week treatment with olmesartan suppressed the elevation of blood pressure and the reduction of phosphorylated eNOS-Ser(1177) in spontaneously hypertensive rats (SHR) resulting to the elevation of eNOS activity.98 Tumor necrosis factor-alpha (TNF-alpha) is known to decrease eNOS expression and is an important mediator of endothelial dysfunction. Kataoka et al. induced a TNF-α decrease in eNOS expression in HUVECsσ. Pretreatment of HUVECs with AT1 receptor blockers (olmesartan or candesartan) restored the TNF-alpha-dependent reduction of eNOS.99
Aliskiren
Aliskiren is the first in a new class of orally effective rennin inhibitors for the treatment of hypertension. Early clinical trials in hypertensive patients showed that this drug provided antihypertensive efficacy comparable to those of the ARBs losartan and irbesartan.100,101 By using a catheter-type NO sensor, Imanishi et al.102 demonstrated that aliskiren administration increases ACh-induced and basal plasma NO concentrations in Watanabe heritable hyperlipidemic (WHHL) rabbits. Moreover, aliskiren/valsartan cotreatment increased the ACh-induced and basal plasma NO concentrations to a significantly greater extent than either aliskiren or valsartan alone. The results of this study could provide an experimental rationale for the combined application of ARBs and renin inhibitors in the treatment of hypertension and related cardiovascular diseases.102
Statins
The most widely used and promising agents in cardiovascular disease, HMG-CoA reductase inhibitors or statins, are renowned for their “pleiotropic” lipid lowering-independent effects. HMG-CoA reductase inhibitors improve endothelial function in many ways. NO bioavailability is increased not only by statins-induced reduction in ROS production but also by direct effects on eNOS enzyme. It has been suggested that statins upregulate eNOS expression and activity103 and prolong eNOS mRNA half-life104 by a post-transcriptional mechanism involving inhibition of geranylgeranylation of Rho GTPase, and stabilization of eNOS mRNA.32,35 Statin-induced activation of phosphatidylinositol 3-kinase – Akt protein pathway also increases NO production and inhibits endothelial cell apoptosis.104 In addition, several statins inhibit endothelial superoxide formation by reducing the activity of NADPH oxidase. This is partly due to the prevention of the isoprenylation of p21phox, which is critical for NADPH oxidase assembly.105 We have recently demonstrated that atorvastatin has a direct effect on vascular superoxide generation in human vein grafts, mainly by reducing NADPH-oxidase activity as a result of the reduction of membrane translocation of p67phox and Rac1, subunits of the enzyme.106 Furthermore simvastatin treatment increases the number of functionally active endothelial progenitor cells as well as SOD activity,107 while, statins also increase GCH1 mRNA expression in endothelial cells and elevate intracellular BH4.108
In addition statins, by suppressing the expression of pro-inflammatory mediators, reduce circulating ADMA plasma levels and improve NO bioavailability, an ability alien to antioxidant vitamins, mainly in diabetic subjects.109
Thiazolidinediones
Many antidiabetic agents are currently in use for treatment of hyperglycemia, however the insulin-sensitizing thiazolidinediones (or TZDs) seem to have additional beneficial effects on vascular wall. Thiazolidinediones are agonists of the peroxisome proliferators-activated receptor gamma (PPAR-γ). PPAR-γ is a nuclear hormone receptor which is involved in the modulation of the expression of several genes that affect glucose and lipid metabolism. In vitro evidence suggests that PPAR-γ is also expressed in vascular smooth muscle cells and endothelial cells.110 One report demonstrated that ciglitazone and a PPAR-γ synthetic ligand can stimulate the release of NO from endothelial cells.110 However Linscheid et al. demonstrated that rosiglitazone reduces intracellular levels of BH4 by inhibiting GTPCH, the rate-limiting enzyme of in BH4 biosynthetic pathway.111
Recent large randomized clinical trials, like the PROACTIVE112 and PERISCOPE113 have illustrated the postulated beneficial effects of TZDs. TZD’s vasoprotective properties also include inhibition of human endothelial cell apoptosis114 and improved stability of eNOS mRNA by inducing eNOS phosphorylation at Ser1177 and promoting heat shock protein 60 – eNOS interaction.114,115 Treatment of hypercholesterolemic rabbits with rosiglitazone reduces oxidative stress, by suppressing iNOS and gp91phox expression, and improves EDD vasodilatation.116
Folates
Folates administration has been long used for the treatment of homocysteinemia.80 Cell culture studies have demonstrated that Hcy reduces eNOS expression in a dose-dependent way, possibly by affecting the activities of DDAH and ADMA.117 In addition, Hcy induces threonine-495-phosphorylation of eNOS a reaction leading to inactivation of the enzyme.118 Moreover Hcy down-regulates eNOS activity by modifying caveolin-1 expression.119 The increased oxidative stress observed in homocysteinemia is responsible for the oxidative degradation of NO.120 In addition, evidence also suggests that vascular tHCy affects O2− generation by modulating NADPH-oxidase activity.121,122
Given these well documented effects of homocysteinemia eNOS expression and activity and NO bioavailability it is sensible to consider that the use of folates can increase eNOS expression and activity as well as NO bioavailability. As we have previously shown, 5-MTHF administration, the circulating form of folic acid, has the ability to prevent peroxynitrite-mediated BH4 oxidation and improve eNOS activity and coupling, mainly by increasing endothelial BH4 bioavailability.78 In addition, we demonstrated that 5-MTHF ameliorates endothelial NO bioavailability and lowers vascular O2− production in human arteries and veins, both in vivo and ex vivo.78 Oral folic acid administration (0.4 mg/day or 5 mg/day) seven weeks before scheduled coronary by-pass grafting, resulted into improved endothelial function and reduced vascular O2− generation due to an improvement of eNOS coupling.79,123 Therefore, homocysteine-lowering treatment with folates may improve eNOS coupling both by reducing intracellular HCy in vascular endothelium and by acting as a direct scavenger of peroxynitrite radicals in endothelial cells. Furthermore, we have recently shown that treatment with folic acid induces a significant improvement of aortic distensibility and reduces indexes of arterial stiffness such as pulse wave velocity.79 Of course treatment with folates can be useful only in the presence of intracellular 5-MTHF deficiency, such as in the presence of 677 TT genotype in the MTHF reductase (MTHFR).81
Polyphenolic antioxidants and angioxidant vitamins
Epidemiological evidence suggests that, dietary polyphenolic antioxidants (fruit, vegetables, wine, grape juice, etc) have a negative correlation with the incidence of cardiovascular disease.124 Even though most polyphenols are only mild antioxidants, some can reduce the activity of pro-oxidative NADPH-oxidases, and others can stimulate antioxidative enzymes and eNOS.125
High doses of vitamin C have been found to improve endothelial function in humans.126–129 Some other studies demonstrated that long-term oral treatment with vitamin C also improved endothelial function in humans.130 Despite all that, long-term epidemiological studies with oral vitamin C treatment failed to support a role for vitamin C in reducing cardiovascular morbidity or mortality.131
Even though vitamin E has antioxidant properties, large studies discourage the use of vitamin E for the prevention of cardiovascular events.132
Endothelin receptor blockade
The 21-amino acid peptide endothelin-1 (ET-1) is the predominant isoform of the endothelin peptide family, which includes ET-2, ET-3, and ET-4. It exerts various biological effects, including vasoconstriction and the stimulation of cell proliferation in tissues both within and outside of the cardiovascular system. ET-1 activates Gi-protein – coupled, 7-transmembrane domain receptors. Five ET receptors have been cloned. Mammals possess ETA133 and ETB receptors.134
Studies in L-NG-nitroarginine methyl ester hypertension suggest that ET-1 is linked to the dysfunction of the
Tetrahydrobiopterin
Despite the well documented role of BH4 in human vessels function, treatment with BH4 was infeasible until recently not only due to the high cost of such treatment, but also because of the high instability of BH4 molecule: BH4 compound readily undergoes aerobic oxidation139 and has a shelf-life of less 8 h at room temperature.140 However a stable tablet formulation of BH4 has been developed recently, wherein the compound retains at least about 98% of the initial amount of (6R)-
Conclusions
It has now become apparent that numerous physiological and pathophysiological stimuli can modulate eNOS expression. Several in vitro and in vivo studies tried to investigate the expression pattern of eNOS gene. Data from the most recent studies suggested that there is a complex regulatory mechanism of eNOS expression, at both transcriptional and post-transcriptional level. More recent studies tried to define the regulatory mechanisms involved in eNOS expression including signaling pathways and cis-trans interaction. Finally, by understanding eNOS molecular biology it is becoming more and more feasible the designation of new therapeutical strategies mainly targeting eNOS expression and eNOS coupling, essential in the treatment of various diseases such as atherosclerosis and coronary artery disease. It is therefore widely believed that eNOS function regulation is a therapeutic target in atherosclerosis since this enzyme not only regulates endothelial NO bioavailability, but it also plays a crucial role in the regulation of vascular redox, affecting in this way multiple redox-sensitive transcriptional pathways in the human vascular wall.
References
Cite this article
TY - JOUR AU - Michael Demosthenous AU - Charalambos Antoniades AU - Dimitris Tousoulis AU - Marios Margaritis AU - Kyriakoula Marinou AU - Christodoulos Stefanadis PY - 2011 DA - 2011/04/21 TI - Endothelial nitric oxide synthase in the vascular wall: Mechanisms regulating its expression and enzymatic function JO - Artery Research SP - 37 EP - 49 VL - 5 IS - 2 SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2011.03.003 DO - 10.1016/j.artres.2011.03.003 ID - Demosthenous2011 ER -