
Chronological changes of aortic and hepatic lesions in apolipoprotein E deficient mice☆
This work was partially supported by an educational grant of the Fundación Florencio Fiorini and by funds from Instituto de Investigaciones Cardiológicas “Prof. Dr. Alberto C. Taquini” (UBA-CONICET).
- DOI
- 10.1016/j.artres.2011.05.001How to use a DOI?
- Keywords
- Apolipoprotein E; Atherosclerosis; Aortic arch; Liver injury; Time-course
- Copyright
- © 2011 Association for Research into Arterial Structure and Physiology. Published by Elsevier B.V. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Background
Apolipoprotein E deficiency impairs lipid metabolism and accelerates atherosclerosis development in a genetically altered mouse model.1 Apo E deficiency is related to the hepatic expression of proinflammatory mediators2 and to accelerated aging in heart, kidneys and liver.3 In relation with hepatic damage, increased susceptibility to develop severe liver injury4 and a 5-lypoxigenase mediated inflammatory mechanism have been reported.5 Yet, despite the wide utilization of apo E-deficient (knock-out, null or (−/−)) mice for multiple purposes, the relationship between hepatic injury and atherosclerotic development is still unclear.6
Aims
This work was conducted in order to explore a possible temporal association between hepatic damage and atherosclerotic evolution in Apo E-deficient mice and to characterize the lesions as well.
Methods
Fifteen newborn apolipoprotein E-deficient male mice (on a C57BL/6J background, The Jackson Laboratory, Bar Harbor, Maine, USA) were fed a standard chow diet. At 18, 20 and 30 weeks of age, consecutive groups of 6 randomly selected mice were euthanized. Procedures were approved by the Animal Care and Use Committee of the University of Buenos Aires. Aortic arch and liver were dissected and immersed in 10% buffered formaldehyde (Formalin 10% buffered solution, pH = 7.0) at room temperature for at least 24 h fixation. After dehydration (graded ethanol series of 50%, 70%, and 100%), tissues were embedded in paraffin blocks and serially cut. Blocks were oriented to allow transverse serial sectioning and localization of the aortic arch. Hematoxylin-eosin and Heidenhain’s Azan trichrome staining were used for histological examination. Acetic orcein stain was used to identify elastic fibers. A qualitative description of plaque histological features included the internal elastic membrane, lipid deposition, plaque inflammation and deposition of calcified material.
Tissue histomorphometry and planimetry were performed using a software-coupled (Image Pro Plus for Windows, v3) Nikon Eclipse E400 microscope. Plaque area and tunica media’ length were measured in digitized magnified sections. Intimal layer and intima/media ratio were considered when atherosclerotic lesions were observed. For liver histological analysis, non-alcoholic steato hepatitis (NASH)7 features were scored (0 = lowest, 4 = highest) including: steatosis, hepatocellular injury, parenchymal inflammation, portal inflammation and fibrosis.
Data were submitted to MANOVA, zero-order and partial bivariate correlations (Pearson’s r coefficient). Alpha level of statistical significance was set at 0.05.
Results
Minute focal accumulations of lipid-laden macrophages were observed at 16 weeks (Fig. 1A). By the 20th week an increased number of macrophages were clustered in globular accumulations. These were covered by a thin fibrous cap and prolapsed in the arterial lumen. The integrity of the internal elastic membrane was spared (Fig. 1B). By the 30th week large acellular necrotic xanthomas formed a fibro-fatty nodule (cholesterol and connective tissue) which extended from the lumen to the internal elastic membrane. Along with this, luminal caliber was largely reduced with thinning and loss of the fibrous cap. Extensive atrophy of the media layer was observed being replaced with plaque components. Internal and external elastic membranes were disrupted and discontinued at the site of plaque development (Fig. 1C).
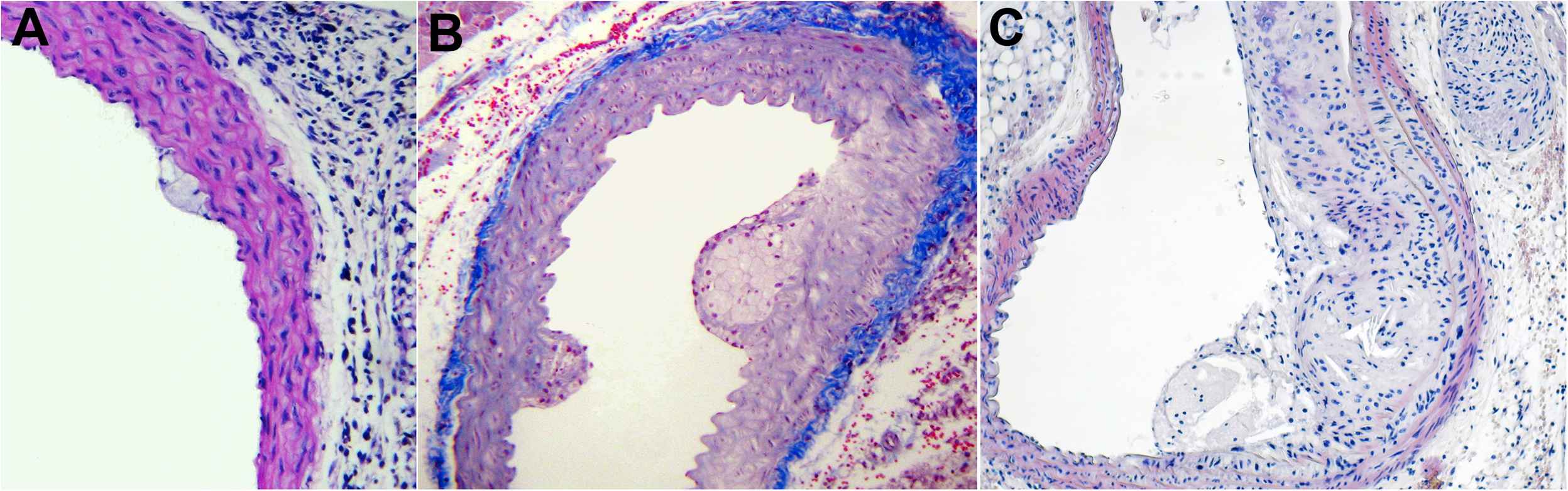
Chronological evolution of atherosclerotic lesion in Apo E-deficient mice. Age in weeks: 16 (A), 20 (B), 30 (C). For description see text.
Plaque area (F1,8 = 10.34, p < 0.012), media thickness (F1,8 = 11.88, p < 0.009) and intima-media ratio (F1,8 = 5.32, p < 0.05) were time-dependent. Increase in plaque area (5-fold, p < 0.0001), stenosis percentage (2.4 fold, p < 0.01) and intima-media ratio (2.6 fold, p < 0.05) along with media thinning (−58% thickness, p < 0.01) were observed from weeks 20–30 (Table 1, Fig. 2).

Plaque area and liver inflammation in Apo E (−/−) mice at 16, 20 and 30 weeks of life.
| Age (weeks) | |||
|---|---|---|---|
| 16 | 20 | 30 | |
| (n) | (4) | (6) | (5) |
| Plaque area (μm2) | – | 20531 ± 6961 | 111536 ± 26131b |
| Stenosis (%) | – | 13 ± 4 | 31 ± 9c |
| Intima (μm) | 11 ± 1 | 139 ± 33a | 164 ± 20a |
| Media (μm) | 56.5 ± 8.0 | 23.9 ± 4.9c | |
| Intima-media | 3.2 ± 0.7 | 8.3 ± 2.2e | |
| (n) | (4) | (4) | (4) |
| Steatosis (0–4) | 3.8 ± 0.3 | 2.8 ± 0.6 | 3.0 ± 0.0 |
| Hepatocellular injury (0–4) | 2.5 ± 0.3 | 2.5 ± 0.7 | 2.8 ± 0.3 |
| Parenchymal Inflammation (0–4) | – | 0.3 ± 0.3 | 1.5 ± 0.3c |
| Portal Inflammation (0–4) | 0.5 ± 0.3 | 0.8 ± 0.3 | 1.8 ± 0.3d |
| Fibrosis | – | – | – |
Data are shown as media ± SE.
p < 0.01 vs. 16 weeks.
p < 0.0001.
p < 0.01.
p < 0.03.
p < 0.05 vs. 20 weeks.
Time course of changes in aorta and liver in apoE deficient mice.
Unlike steatosis or hepatocellular injury, liver parenchymal inflammation (F2,9 = 13.29, p < 0.002) and portal inflammation (F2,9 = 6.30, p < 0.02) were time-dependent and increased by 5-fold (p < 0.01) and 2-fold (p < 0.03) respectively from weeks 20–30 (Table 1). Fibrosis was not observed at any time (Fig. 3).
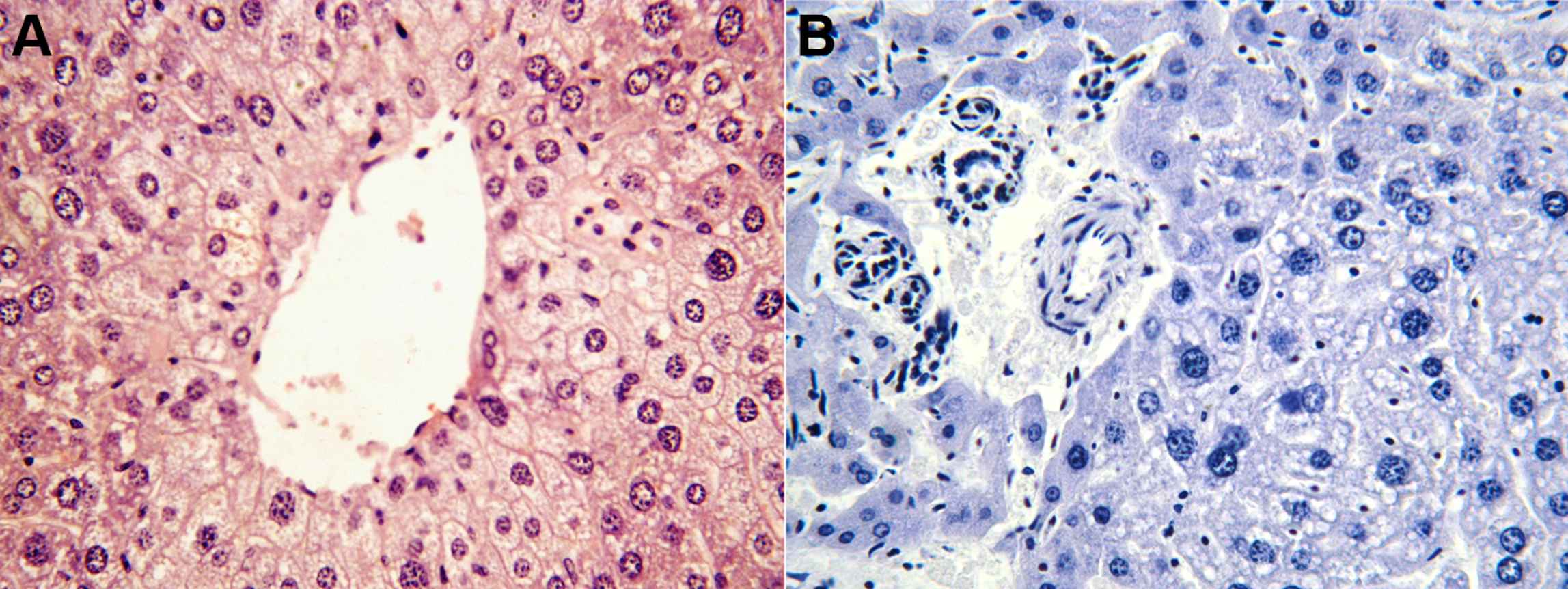
Hepatic steatosis (left) and parenchymal inflammation (right) in 30 weeks’ Apo E-deficient mice.
Interestingly not only time accounted for 62% of plaque area variation and 53% of changes in liver parenchymal inflammation but both variables mirrored each other’s time-course as well (Fig. 2).
Conclusion
High variability of plaque area and liver parenchymal inflammation found around 20 weeks could not be explained by time and exceeded interindividual variability observed at any other time. An evolving period characterized by rapid changes might alternatively explain high variability in plaque area and parenchymal inflammation observed at this time. The estimated half-time for severe lesions development as predicted by a sigmoidal data fitting equation (∼20.4 weeks) is in agreement with previous reports showing full development of lesions around 40 weeks.8
A complex interplay of adhesive cellular interactions, chemotactic factors, proinflammatory chemokines and growth-regulatory molecules participates in atherogenesis.9 Knowledge of the temporal course of putative atherosclerosis-related inflammation mediators across life-span might help to identify key candidates responsible for atherosclerosis evolution in this mouse model.
Atherosclerotic evolution might be associated with the progression to a severe inflammatory form of liver damage in apolipoprotein E-deficient mice. Present findings may suggest that a series of rapid and transient changes might take place around 20 weeks of life. These might be critical in determining the progression of tissue damage in this mouse model of atherosclerosis.
References
Cite this article
TY - JOUR AU - Matilde Otero-Losada AU - Santiago McLoughlin AU - Gastón A. Rodríguez-Granillo AU - Angélica Muller AU - Graciela Ottaviano AU - José Milei PY - 2011 DA - 2011/07/02 TI - Chronological changes of aortic and hepatic lesions in apolipoprotein E deficient mice☆ JO - Artery Research SP - 109 EP - 111 VL - 5 IS - 3 SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2011.05.001 DO - 10.1016/j.artres.2011.05.001 ID - Otero-Losada2011 ER -