
Immune Thrombocytopenia: Are We Stuck in the Mud or Is There Light at the End of the Tunnel?
- DOI
- 10.2991/chi.d.190805.001How to use a DOI?
- Keywords
- Leukemia; Platelets; Diagnosis; Autoimmunity
- Abstract
The differences in diagnosis and management between immune thrombocytopenia (ITP) and leukemia are striking. Leukemia diagnosis and management have evolved substantially over the past 30 years and are now relatively precise. The tendency for leukemic cells to be the overwhelming majority of circulating and/or bone marrow cells certainly continues to facilitate developments, as does improved molecular assessment. Furthermore, randomized controlled clinical trials of competing regimens in well-defined populations have advanced treatment as well. Currently, ITP diagnosis and management depend very much on the experience and preferences of the hematologist. There are no unequivocally useful molecular tests, no agreement on which testing needs to be performed, and no consensus on treatment. Future studies using advanced techniques would ideally change this over time but, thus far, progress in ITP has been slow. However, the increasing ability to do single-cell DNA and RNA studies and flow cytometric dissection of small populations of cells could radically change the approach to ITP if critical distinctions were uncovered.
- Copyright
- © 2019 International Academy for Clinical Hematology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
In this age of molecular diagnosis, prognostication and application of targeted therapies, immune thrombocytopenia (ITP) is a throwback to the way medicine was practiced 30 years ago. The most obvious way to highlight the issues is to compare the current approach in ITP with that in leukemia (see Figure 1).
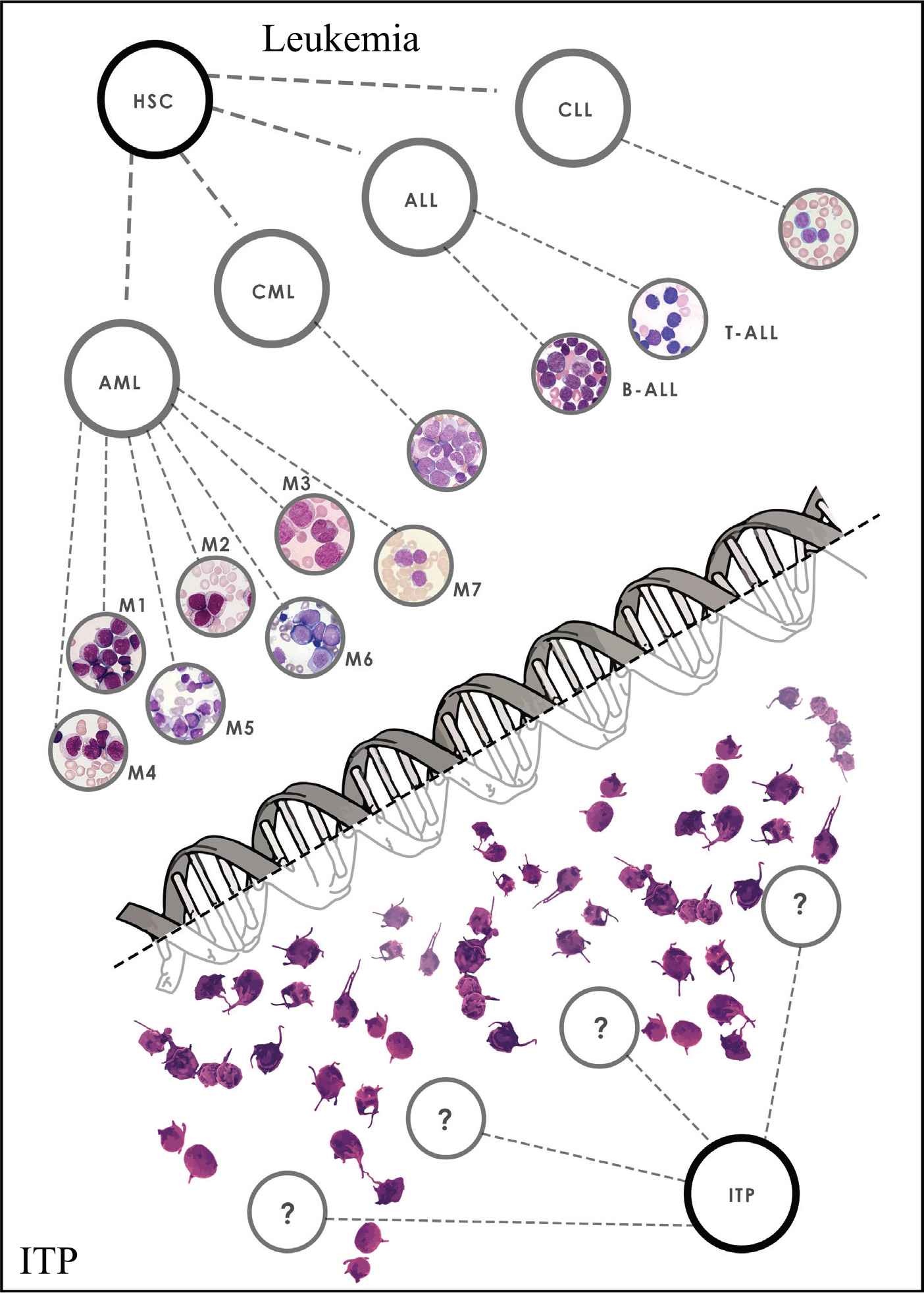
The staggering differences between diagnosis and management of immune thrombocytopenia and leukemia. Figure Citations [35–39] Leukemia: (i) Quick diagnosis, (ii) Molecular classification, (iii) Accurate prognostic indicators, (iv) Predefined treatment, (v) Predefined alternative treatments. ITP: (i) Diagnosis of exclusion, (ii) No diagnostic testing, (iii) No molecular testing, (iv) No predefined treatments, (v) No predefined alternative/escalation treatments.
ITP and leukemia, whether in children or in adults, are complex diseases. Even in the best-case scenario of lymphoblastic leukemia in childhood, with a 90+ percent estimated survival, no one would choose to have leukemia and, certainly not, to have leukemia instead of ITP. Current estimate for mortality secondary to an intracranial hemorrhage in children with ITP is something around 1 in 200–500 children. Morbidity can be a result of concurrent disease, that is, hypothyroidism or chronic fatigue, and other features of impaired quality of life. Fortunately, most treatment-related morbidity in a well-managed patient is transient and not lasting. There are striking differences in the management of leukemia and ITP that have important impacts on the diagnosis and treatment (and, of course, the outcomes) of the two diseases.
Thirty years ago, leukemia would usually be diagnosed when the white cell count was high (or low) and the smear would look abnormal, showing blasts. At that point, an experienced morphologist would review the smear and discuss which type of leukemia this looked like, prompting examination of bone marrow morphology and histochemistry to identify which type of blasts these were, that is, myeloid or lymphoid. This was the era of the French–American–British (FAB) classification [1]. Cytogenetics to identify Philadelphia chromosome t(9;22) in chronic myeloid leukemia (CML) and the t(15;17) translocation for Acute Promyelocytic Leukemia (APL) could be regularly obtained and, soon thereafter, flow cytometry was routinely performed in all cases. Once flow cytometry was established, it began to be performed on peripheral blood, so that leukemia could be confirmed and its type identified within 4–12 hours of the patient's arrival in the Emergency Room (ER), even prior to performing bone marrow examination. Furthermore, both the quality of the flow cytometric instruments and the number of different monoclonal antibodies available to identify cell surface antigens increased substantially. These two developments created the ability to simultaneously use higher number of color flow channels and, thus, to have better resolution of cell populations down to very small percentages. Since then, many different genetic abnormalities have been uncovered, especially in cases of AML, and, as a result, there are many different subtypes of leukemia identified beyond acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), acute myeloid leukemia (AML), APL, and chronic myeloid leukemia (CML) [2–4]. In addition, for many of these different types of leukemia, there are specific targeted therapies, for example, FLT3-ITD [5,6] and IDH-1 and 2 [7,8] AML and, of course, for Philadelphia chromosome ALL [9], CML [10,11] and APL [12]. These treatments increase not only the chance of achieving remission but also of making it long lasting. In the model case of APL, newer targeted treatments result in the side effects and long-term toxicities of conventional chemotherapy being dramatically decreased, while simultaneously providing a very high rate of cure. Accordingly, when someone is diagnosed with leukemia in 2019, the specific subtype is promptly identified, and the patient is treated according to that subtype.
Once leukemia is diagnosed, typed and risk group assessed, the treatment protocol is decided upon. This is planned out until the end of therapy, including long-term follow-up. If the patient does not respond, or responds but relapses, or suffers important toxicity, then the treatment plan is modified. However, if this happens, the modification is something already considered and incorporated into the protocol at diagnosis. The “salvage protocol,” especially if the need to change treatment occurs one or more years after diagnosis, may be different, but it will generally be the next best treatment. Whatever this second protocol is, for example, for lack of response, toxicity, or relapse, it too will be fully scripted out. Therefore, leukemia treatment is relatively inflexible and not susceptible to being changed, for example, according to the patient's preference for tolerability issues.
Laboratory investigations of newly diagnosed patients with leukemia tend to be somewhat uniform across centers, even though there are occasional differences. For example Pro Thrombin Time and Partial Thromboplastin Time (PT–PTT), immunoglobulin levels, liver and kidney tests, blood type and anti-red cell antibody screen, and testing for certain viruses, for example, cytomegalovirus (CMV), are performed in the great majority, if not all patients.
Let us now contrast diagnosis and treatment of leukemia with diagnosis and treatment of ITP. The entire approach is very different because there is so much less specific knowledge about ITP. At the simplest level, ITP is diagnosed as a platelet count <100,000/uL without an explanation. A more extensive description would be that primary ITP is a syndrome characterized by isolated thrombocytopenia, normal red cell and white cell counts and differentials, no hepatosplenomegaly or lymphadenopathy, and no apparent underlying cause of the thrombocytopenia [13]. Especially in children, ITP at presentation is usually an obvious, uncomplicated diagnosis. In adults, however, it may be more difficult. An elderly adult with acquired thrombocytopenia may be sick, be on multiple medications, have multiple comorbidities, be febrile, have transaminitis, and, potentially, other medical issues. In such a patient, the absence of a specific test to confirm ITP is a big liability preventing clarification of the cause of thrombocytopenia.
The general rubric is to do a complete blood count (CBC) with differential, platelets, and review of the smear. No additional testing is required if a patient has what appears to be typical ITP: isolated thrombocytopenia with large platelets, normal red and white cell counts with normal differential, no hepatosplenomegaly or lymphadenopathy, no underlying cause of thrombocytopenia, and absence of relevant medical and family history. Currently, there is no routine testing of “primary ITP” to distinguish it from secondary types of ITP. Platelet antibody testing is done in very few centers as part of the routine evaluation of ITP, because of its lack of specificity [14]. Depending upon the lab performing the test, sensitivity may be variable. In neither case is it diagnostic, because specificity is low. A poll of 10 medical advisors of the Platelet Disorders Support Association revealed that they would do additional testing for various disorders, that is, measurement of immunoglobulin levels or liver tests, but there was absolutely no consistency among any two of the 10 responders, that is, no two would recommend exactly the same set of tests. Furthermore, only a small minority would do platelet antibody testing. Therefore, there is no standard whatsoever as to which, if any, additional tests to do beyond a CBC.
Perhaps, most importantly, there is no current evidence that specific molecular abnormalities need to be considered. In particular, there is no gene mutation identified in patients with ITP, the finding of which would confirm the diagnosis, nor are there genetic tests that unequivocally point to which treatment to use or to avoid. Certainly, there are individual studies which have suggested that one or another test might be useful. However, none have been confirmed in large studies, and none have been repeated and found to be unequivocally correct and useful in a major fraction of patients. The only tests which are helpful if “positive” are those that identify a specific type of either secondary ITP or inherited thrombocytopenia; however, such results each are only found in 1%–3% of cases. In these few patients, their identification may suggest which treatment approach to pursue. Examples include common variable immunodeficiency (CVID) [15,16] unsuspected CMV infection [17], and autoimmune lymphoproliferative syndrome (ALPS) [18], all of which can be managed by specific approaches which are not the same as those that would be considered for primary ITP. However, even if an extensive panel of testing for secondary and inherited ITP is performed, the percentage of “ITP cases” in which an etiology will be identified is relatively small, that is, at most one in five cases [19].
As if this were not bad enough (being unable to meaningfully distinguish among the great majority of cases of ITP), it is practically impossible to know when a case of ITP is drug-induced [20]. While changing all concomitant medications is now more possible than it used to be, given the greater availability of equivalent alternatives, in a given patient it nonetheless may be difficult. Also, it is quite possible that a drug instigated the ITP but is not required for its perpetuation. A given case of “drug-induced thrombocytopenia” may even be caused by a drug metabolite, making laboratory testing of the drug (incubated with the patient's platelets and serum) falsely negative. Whether this form of ITP (drug-induced ITP) behaves differently from primary (idiopathic ITP) is unknown, in large part because its incidence and natural history are so unclear.
In summary, there is currently very limited capability to provide a specific diagnosis of ITP that meaningfully distinguishes one case from another regarding their prognostication, availability of biomarkers to inform on treatment options, and clarification of who is at risk for serious bleeding. The current definitions are largely based on clinical factors (i.e., severe, acute, chronic, recurrent, refractory), not specific laboratory testing. Unlike leukemia, ITP is not routinely divided into subgroups, each with a different, specific treatment protocol, and prognosis. In general, all cases of ITP are relatively similar, and the primary distinction is eventually based upon the course and response to treatment, if any is administered.
Similarly to the lack of definition for diagnosis, there is limited consensus even for first-line therapy. While there is good general agreement on which treatment to use in adults with newly diagnosed ITP, if treatment is required (i.e., steroids), there is no clear agreement nor standard of practice as to whether dexamethasone (Dex) or prednisone is preferable [21]. A meta-analysis has surprisingly shown a lack of difference in long-term outcome between the two options (prednisone vs Dex), notwithstanding the “presumed” curative effects of Dex. However, the analysis suggested that high dose dexamethasone would bring the count up faster and steroids might have less toxicity when they are used for a very short course, even at higher doses. If Dex is chosen, there is the question of whether more than one 4-day cycle should be given and at which intervals. If regular cycles are given (i.e., at 2-week intervals), this might help “cure” the ITP but also might expose certain patients to treatment they do not need. On the other hand, if additional cycles of dexamethasone are withheld until the count falls, this approach may vitiate the chance of a curative effect [22]. If prednisone is chosen, exactly which dose and how long to continue it remain wide open. The soon forthcoming American Society of Hematology (ASH) guidelines will strongly suggest that the initial course of prednisone should not exceed 6 weeks.
Similarly, there is no consensus in practice on when to use Intra Venous Immuno Globulin (IVIG), neither when to use it instead of steroids nor when to use it with steroids. Therefore, even with first-line therapy, where there would be some unanimity in using steroids, whether or not to add IVIG remains in the eye of the beholder. These choices are not driven by clear clinical data or by individual patient factors, but rather by individual physician preference, based on familiarity, financial factors like earlier hospital discharge if IVIG is used, and so on. Even platelet transfusions are used surprisingly commonly, again reflecting on the difficulty in being certain of the diagnosis and of the risk of bleeding although, admittedly, there are other reasons, such as physician's anxiety.
Also important, there is no set dogma that once a certain treatment is started, the treatment needs to be continued in a certain way. What this means is that, unlike in leukemia, if the physician and the patient decide to start treatment with a certain plan, the patient (or the physician) could say “I don't like this treatment” for whatever reasons. It could be side effects, the route of administration or the expense, among many other possibilities. There would be no imperative reason why the physician would say “I am sorry, but you have to continue with this treatment.” Instead one could say “All right. It might be better for you to go ahead with the treatment we started, but we can go ahead and change to plan B if you so desire.” All together, this indicates that the contrast of diagnosing and managing leukemia as compared to diagnosing and managing ITP is staggering. This example also points to the fact that the diagnosis and treatment of ITP needs to be rediscussed frequently with the patient. Especially at and soon after diagnosis, the patient may take time to assimilate disease information and, thus, may change his or her mind, necessitating considerable time spent with the patient on multiple visits. An additional conundrum is that, at diagnosis, the patient is carefully instructed about the risk of intracranial hemorrhage. Afterwards, once a successful treatment is initiated or the patient improves, the physician counsels the patient reassuringly that this will not happen. For patients with leukemia, the fixed treatment path minimizes the need for frequent discussions: you either are eventually cured or you are not.
It is also worth pointing out that, in ITP, there is relatively little information whereby one treatment is compared to another. This is especially true in second-line therapy. In children, there are a number of comparative studies of first-line treatments. These have primarily explored IVIG compared to prednisone, but have also included IV anti-D and “watch and wait.” There are fewer adult studies and fewer patients enrolled on them [21]. IVIG administration strategies have been compared, and prednisone dosing and comparison to dexamethasone have been pursued.
Comparisons of forms of steroids (dexamethasone to prednisone) are relatively numerous, perhaps not only for reasons of cost, but also because patients at diagnosis are the most numerous and thus easiest to study. Interestingly, unlike leukemia in which a study demonstrating superiority of one regimen over another is likely to rapidly move to becoming the “new” standard of care, ITP is very different. Changes in treatment move more slowly, and seminal papers may or may not change practice. One reason for this is the difficulty in which many countries can access a specific (read: expensive) treatment, that is, Thrombopoietin (TPO) agents, which tends to result in the following of a set pathway, according to ease of availability, rather than of evidence-based analysis of clinical data and incorporation of recent results. Another reason is that the “seminal” studies in ITP are not randomized controlled trials.
There is virtually no consensus whatsoever on second-line therapy. In general, the “defining” studies have been carried out almost exclusively as single-arm treatment or in comparison to placebo, not in comparison to another treatment option. There are randomized placebo-controlled trials to demonstrate the primary efficacy of certain treatments (especially the TPO agents and fostamatinib) [23,24], and these trials illustrate what the therapeutic effects and side effects are. However, no randomized trials including splenectomy have ever been performed. The only sufficiently sized randomized rituximab trial compared it with both arms receiving low dose prednisone [25]. There was an important early effect but it disappeared after 1.5 years. There have been at least nine large TPO trials for ITP, but each only had comparison to placebo [26]. Thus, it is very hard, if not impossible, to make clear conclusions about the relative efficacy of treatments, given the potentially different patient populations included in different trials. We are attempting to solve this massive evidence gap by doing a medical decision analysis to provide the best possible comparison of second-line treatment strategies. However, this will likely not be as accurate as a true randomized comparative trial.
Currently, in choosing treatment for a given patient, we are forced to rely on certain individualization factors in ITP which appear to be important. For example, it appears that rituximab works better in young women [27,28]. This, and having CVID are, by far, the best predictors of response to rituximab. Of note, these factors seem to be prognostic as to the potential “cure” post rituximab, but they do not correlate as well with the initial early response. Therefore, we cannot predict who will have a long lasting or definitive remission to rituximab, since a relapse may happen up to a year after treatment. Such a long progression-free survival, nonetheless, is quite beneficial to the patient.
In addition to not knowing which treatment to choose, there are many other issues to consider. One is what happens if somebody is on a TPO agent and seems to require it in order to maintain an adequate platelet count. Is this an acceptable long-term strategy (even leaving out any considerations of expense)? How do you know when the patient has improved and might not have a normal but rather at least an adequate platelet count even without further treatment with thrombopoietic agents? Data from the long-term studies have suggested that the rate of toxicity decreases over time, that is, thrombosis is most prominent in the first year, but the risk certainly never goes down to zero. Another issue with rituximab, is what if somebody responds and relapses? How often can it be given? Will patients eventually become hypogammaglobulinemic? Will they stop responding? Is three courses of four infusions of rituximab the limit at which point one needs to change treatment? And 20 years after the initial ITP study, what is the optimal (lowest effective) dose of rituximab? And what about long-term side effects? Progressive multifocal leukoencephalopathy is very very rare and even lasting hypogammaglobulinemia is very uncommon; however, even if uncommon, these side effects do occur. If a case of ITP is one of the rare ones in which a lymphoma will develop later, then it is possible that rituximab will mask it but is very unlikely to induce it [29]. In addition, it is unknown whether initial use of rituximab would have any effect on the long-term outcome of the lymphoma treatment.
Splenectomy is the agent for which analysis of long-term outcomes becomes very difficult. It seems clear that the risk of overwhelming postsplenectomy sepsis continues indefinitely, and the complication can occur 10, 20, 30 years after splenectomy [30–32]. Similarly, the risk of venous thrombosis has been shown to be increased in ITP, in general, but appears to be even higher in postsplenectomy patients, with a 1.5 fold increased risk of stroke [33]. Over time, these side effects are important enough to potentially affect the initial choice of treatment. There is as of yet no evidence of an increased rate or frequency of infections after splenectomy, but overwhelming postsplenectomy sepsis and infection with intracellular organisms, for example, malaria, babesia, dengue may be very difficult to treat. At least one study reported increased incidence of myocardial infarction, but this has not been confirmed in other studies. Moreover, there is no evidence of development of pulmonary hypertension after splenectomy for ITP, unlike what is seen in splenectomized patients for hereditary spherocytosis or other hemolytic anemias. However, the evidence in any direction regarding the long-term presence or absence of effects remains less certain than it could be, because of the inherent difficulties in gathering very long-term data in large numbers of patients.
Another uncertainty in the choice of treatment involves the role of fostamatinib. Fostamatinib is a Syk inhibitor that was licensed in April 2018 in the United States for chronic ITP [23,34]. The labeling in the package insert is virtually identical to that of TPO agents. In the absence of comparative data, it is not clear if fostamatinib should be reserved for patients who do not respond to the TPO agents (over 1/3 of the 70 TPO-failure patients responded to fostamatinib). Single-arm data suggest that the antithrombotic effects of fostamatinib may be especially useful in cases of ITP in which there is an increased risk of thrombosis. However, how to define which type or level of risk of thrombosis would suggest use of fostamatinib over another agent is unclear; fostamatinib does have tolerability issues.
As newer agents are developed, for example, the FcRn or the BTK inhibitors, it remains uncertain as to when they are indicated. The good news for patients with ITP is the relatively large number of agents that can be used, which means that there is a wide variety of choices for first or even second-line treatments. However, there is a lack of a data-driven protocol to select which ITP patient should be treated with which agent. A similar lack of evidence exists in selecting which agent to use if the first agent failed, or which patients may benefit from a change of second-line therapy or from combination therapy. It would be a very important benefit if individualized factors that affect treatment outcomes could be better understood. Studies with longer follow-up and good definition of eligibility criteria (enrolled patient population) would be very helpful.
The current approach tends to be “penny-wise” with short-term comprehensive evaluation which perpetuates lack of certain important information and, thus, may be “pound foolish.” Specifically, it may look at short-term responses rather than “cure” or, at least, long-term safety and efficacy. Much of this is drawn from “pharmaceutical wisdom” intended not to circumscribe any potential market: “let's only study and pursue what we have to study and pursue.” In patients with leukemia and cancer, the field is so competitive, the stakes so high, and the treatments so expensive, that specific treatments are marketed for limited subsets of a given disease type, for example, targeted therapy for a specific genetic aberration.
Thus far, this article has focused on what is ill-defined about diagnosis and management of ITP. But there are a number of very good things about ITP. First, in general, the mortality is very low and is almost comparable to that of a person who does not have ITP. In the medical decision analysis alluded to previously, there is a very small difference in lifespan (less than 1 year, i.e., 80 versus 81 years) between a 40-year-old woman with ITP and one without ITP. ITP can be mild enough so that there is considerable debate (especially in children) about whether any treatment should be administered. If treatment is to be given, there are manyfrom which to choose. Even if comparative information for the treatments does not exist, there is considerable information available on each one, including substantial safety data on all licensed treatments. Furthermore, new ones continue to be developed and considerable research continues apace.
In the final analysis, it is clear that no one would choose to have leukemia if they could have ITP instead. While many things about leukemia are very well defined, it is nonetheless a disease that can be fatal, and one for which the treatments in no way, shape, or form resemble health options. In 1980, treatment choices for patients with ITP were primarily prednisone versus splenectomy. Subsequently, in 1981, IVIG was introduced, followed by rituximab, the TPO agents and, most recently, fostamatinib. Currently, FcRn blockers and BTK inhibitors are being developed with promising intial results. The introduction of all of these agents and others has created a bewildering array of “riches” which, in turn, have encumbered the selection process. As studies continue, and techniques of molecular biology advance and are applied to ITP, the sophistication of the analysis now used in assessment of leukemia should gradually enter the world of ITP and completely change its landscape.
CONFLICT OF INTEREST
Oriana Miltiadous none
James Bussel: consultant and/or member of advisory boards: Amgen, Novartis, Dova, Rigel, Momenta, Argenx, UCB, Regeneron, and RallyBio
Speaker: Novartis, Shenyang Biologics
Honoraria: Up to Date
AUTHORS' CONTRIBUTIONS
J.B. and O.M. wrote and edited the manuscript, and agreed on the final version for submission.
ACKNOWLEDGMENT
The authors gratefully thank Iris Miltiadous for help with the figure.
REFERENCES
Cite this article
TY - JOUR AU - James Bussel AU - Oriana Miltiadous PY - 2019 DA - 2019/08/12 TI - Immune Thrombocytopenia: Are We Stuck in the Mud or Is There Light at the End of the Tunnel? JO - Clinical Hematology International SP - 173 EP - 179 VL - 1 IS - 4 SN - 2590-0048 UR - https://doi.org/10.2991/chi.d.190805.001 DO - 10.2991/chi.d.190805.001 ID - Bussel2019 ER -