
KCa3.1 Inhibition Decreases Size and Alters Composition of Atherosclerotic Lesions Induced by Low, Oscillatory Flow

- DOI
- 10.2991/artres.k.210202.001How to use a DOI?
- Keywords
- Kcnn4; REST; NRSF; oscillatory flow; carotid artery; atherosclerosis
- Abstract
Low, oscillatory flow/shear patterns are associated with atherosclerotic lesion development. Increased expression of KCa3.1 has been found in Vascular Smooth Muscle (VSM), macrophages and T-cells in lesions from humans and mice. Increased expression of KCa3.1, is also required for VSM cell proliferation and migration. Previously, we showed that the specific KCa3.1 inhibitor, TRAM-34, could inhibit coronary neointimal development following balloon injury in swine. Atherosclerosis develops in regions with a low, oscillatory (i.e. atheroprone) flow pattern. Therefore, we used the Partial Carotid Ligation (PCL) model in high-fat fed, Apoe−/− mice to determine the role of KCa3.1 in atherosclerotic lesion composition and development. PCL was performed on 8–10 week old male Apoe−/− mice and subsequently placed on a Western diet (TD.88137, Teklad) for 4 weeks. Mice received daily s.c. injections of TRAM-34 (120 mg/kg) or equal volumes of vehicle (peanut oil, PO). 1-[(2-chlorophenyl) diphenylmethyl]-1H-pyrazole (TRAM-34) treatment reduced lesion size ~50% (p < 0.05). In addition, lesions from TRAM-34 treated mice contained less collagen (6% ± 1% vs. 15% ± 2%; p < 0.05), fibronectin (14% ± 3% vs. 32% ± 3%; p < 0.05) and smooth muscle content (19% ± 2% vs. 29% ± 3%; p < 0.05). Conversely, TRAM-34 had no effect on total cholesterol (1455 vs. 1334 mg/dl, PO and TRAM, resp.) or body weight (29.1 vs. 28.8 g, PO and TRAM, resp.). Medial smooth muscle of atherosclerotic carotids showed diminished RE1-Silencing Transcription Factor (REST)/Neural Restrictive Silencing Factor (NRSF) expression, while REST overexpression in vitro inhibited smooth muscle migration. Together, these data support a downregulation of REST/NRSF and upregulation of KCa3.1 in determining smooth muscle and matrix content of atherosclerotic lesions.
- Copyright
- © 2021 The Authors. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
Despite lipid lowering and emerging anti-inflammatory agents, atherosclerosis remains the leading cause of death in both men and women in the United States [1,2]. An estimated 15 million Americans >20 years of age have Coronary Heart Disease (CHD) while each year ~635,000 Americans have a new coronary attack and ~300,000 have a recurrent attack [1]. Atherosclerosis is an inflammatory, proliferative disease that develops over decades, and includes the involvement of numerous cell types including endothelial cells, smooth muscle cells, fibroblasts, macrophages, T-cells, and B-cells as well as platelets. The intermediate-conductance Ca2+-activated K+ channel (KCa3.1) is expressed in all of these cell types, and contributes to T-cell, B-cell, fibroblast, and vascular Smooth Muscle Cells (SMC) proliferation; as well as the migration of Vascular Smooth Muscle (VSM) and macrophages and platelet coagulation [3–7]. Accordingly, increased expression of the KCa3.1 has been observed in smooth muscle, macrophages and T-cells in atherosclerotic lesions from humans and mice [6], while treatment of Apoe−/− mice with the KCa3.1 inhibitor, TRAM-34, produced smaller lesions in the aortic root [6]. Synthetic, proliferating VSM cells increase expression of KCa3.1, such that it becomes the dominant K+ channel [7,8]. Although the cell type-specific relative contribution of KCa3.1 activation during atherosclerosis has not been determined, upregulation of KCa3.1 has been observed in neointimal smooth muscle cells of balloon-injured rat carotid arteries [6,9]. In addition, we showed that acute administration of TRAM-34 during coronary angioplasty could inhibit lesion development in a swine model of coronary restenosis that is predominantly a smooth muscle proliferative response [4]. Thus, KCa3.1 activation in VSM likely contributes to plaque formation.
Atherosclerotic lesions are known to develop predominantly in regions of disturbed flow characterized by low, oscillatory shear in humans and animal models of atherosclerosis [10,11]. Partial ligation of the mouse carotid artery reduces blood flow and shear >90%, producing disturbed flow with characteristic low and oscillatory wall shear stress [12–15]. When partial carotid ligation is performed in a mouse model of atherosclerosis, e.g. the Apoe null mouse on a high cholesterol diet, complex atherosclerotic lesions develop in the carotid artery proximal to the ligation inducing endothelial dysfunction at 1 week and advanced lesions by 4 weeks [13,16].
In the early stages of atherosclerosis, arteries enlarge in relation to plaque area (i.e. outward or positive remodeling) to preserve lumen until lesion area exceeds ~40% of vessel area [17], a.k.a. Glagov’s phenomenon. In human atherosclerotic coronary arteries, lumen areas are maintained in regions of low shear despite an increase in plaque size by an increase in vessel area, i.e. the external elastic lamina area [18]. Approximately 60% of arteries compensate appropriately, while others fail to remodel or show excessive expansive outward remodeling which increases risk for plaque rupture. Smooth muscle de-differentiation, migration and matrix remodeling are essential to vascular remodeling. Whether KCa3.1 activation plays a role in compensatory remodeling during atherosclerosis is unknown. Thus, we sought to determine the role of KCa3.1 in atherosclerosis development and compensatory remodeling in a model of disturbed flow.
2. MATERIALS AND METHODS
2.1. Ethics Statement
Experimental protocols were approved by the University of Missouri Animal Care and Use Committee and in accordance with the “Principles for the Utilization and Care of Vertebrate Animals used in testing, Research and Training.”
2.2. Experimental Animals
Male Apoe−/− mice (B6.129P2-Apoetm1Unc/J) were purchased from Jackson Laboratory (Bar Harbor, MN, USA) and were fed ad libitum with standard chow diet until surgery at 8–10 weeks of age. Partial carotid ligation was performed as previously described [13,19]. Partial carotid ligation involves ligation of three of the four branches of the Left Common Carotid Artery (LCCA: see Figure 1), leaving only the superior thyroid branch patent. Mice were anesthetized by intra-peritoneal injection of xylazine (10 mg/kg) and ketamine (80 mg/kg). The ventral side of the neck was epilated, disinfected and a ventral midline incision was made to expose the left carotid artery. The Internal Carotid Artery (ICA), External Carotid Artery (ICA) and occipital artery were ligated with 6-0 silk suture. The incision was approximated and closed with surgical glue. Following surgery, mice were switched to a Western diet (0.2% cholesterol, 42% kcal from fat; TD.88137, Teklad Diets, Envigo, USA) and received daily subcutaneous injections of TRAM-34 (120 mg/kg; n = 12) or equal volumes of vehicle (peanut oil, PO; n = −10). Four to five weeks post-surgery, mice were anesthetized and perfusion fixed at 80 mmHg with 10% neutral-buffered formalin. The LCCA (distal, mid and proximal) and Right Common Carotid Artery (RCCA) were dissected, fixed and embedded in paraffin for subsequent histology.
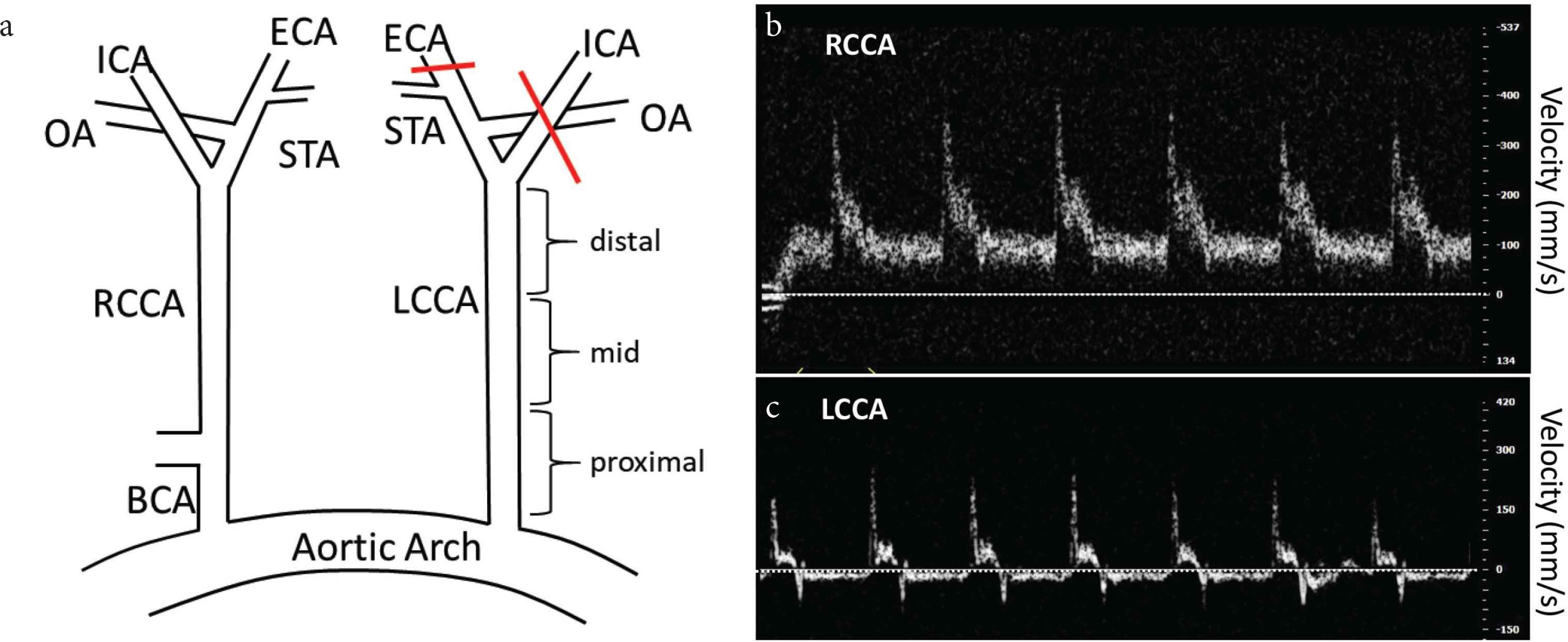
(a) Schematic of partial-carotid ligation. Red lines indicate ligation. (b) and (c) Flow velocity in RCCA and LCCA 2 days post-surgery. Horizontal line = 0, velocity increments = 100. Note reduced, oscillatory (retrograde/antegrade) flow in LCCA.
2.3. Doppler Flow Measurement
Doppler velocity signals were measured on both LCCA and RCCA arteries using a high-resolution ultrasound (Vevo 2100, VA Small Animal Imaging Core). After induction with 3–4% isoflurane in a stream of oxygen anesthetized mice were placed supine on a thermostatically controlled heated platform (Indus Instruments Mouse Monitor-S vital signs monitoring system) set to 40–42°C and limbs were taped to ECG electrodes embedded in the surface of the platform. Isoflurane level during the rest of the procedure was maintained at 1.5–2% and ECG, HR, and respiration rate was continuously monitored. The probe was placed on the left and right sides of the neck, close to the carotid entrances and oriented toward the flow at a 45° angle, to detect flow velocity. Doppler spectra were recorded for offline analysis.
2.4. Immunohistochemistry
Immunohistochemistry was performed as previously described [4]. Sections were incubated with avidin–biotin two-step blocking solution (Vector SP-2001, Vectorlabs, USA) to inhibit background staining and 3% hydrogen peroxide to inhibit endogenous peroxidase. Non-serum protein block (Dako X909, Agilent, USA) was then applied to inhibit non-specific protein binding. Sections were incubated at 4°C overnight with primary antibodies SMα A (1:200, Dako catalog #M0851), fibronectin (1:500, Santa Cruz catalog #8422) and REST/NRSF (1:20,000, gift from Dr. David Beech). After washing, sections were incubated with biotinylated secondary antibody in phosphate-buffered saline containing 15 mM sodium azide and peroxidase-labeled streptavidin (Dako LSAB+ kit, peroxidase, K0690, Agilent, USA). Diaminobenzidine (Dako, Agilent, USA) was applied 5 min for visualization of the reaction product, sections were then counterstained with haematoxylin, dehydrated, and coverslipped. Images of the sections were obtained using an Olympus BX61 photomicroscope and Spot Insight Color camera (Diagnostic Instruments, USA). The relative area and mean density of positive staining were determined for each section of interest utilizing ImagePro Plus (Media Cybernetics, Rockville, MD, USA).
2.5. Morphology
Sections were obtained from the proximal, mid and distal portions of the LCCA. Morphometric measurements were obtained on spatially calibrated images of Verhoeff-Van Gieson (VVG) stained sections using standard planimetry and NIH Image J software (Bethesda, MD, USA). Intimal area was defined as the area between the Internal Elastic Lamina (IEL) and the luminal border of the artery while the area between the IEL and External-Elastic Lamina (EEL) was referred to as the medial area. Intima-Media ratio (I/M) was calculated as the ratio of intimal-over medial-areas. Percent stenosis was defined as intimal area/IEL area.
2.6. Blood Analysis
Total cholesterol content was determined by Comparative Clinical Pathology Services, LLC (Columbia, MO, USA) from serum collected the day of euthanasia.
2.7. Cell Culture
Primary cultures of Mouse Aortic SMC (MASMC) from WT control B6 (C57BL/6, JAX) and Kcnn4−/− (B6; 129S1-Kcnn4tm1Jemn/J, JAX) mice were isolated from the thoracic, following removal of adventitia [20]. Cells were plated at 1.5 × 104 cells/cm2 in DMEM/F-12 media (Thermo Fisher 11320-033, Waltham, MA, USA) containing 100 U/mL pen/strep, 1.6 mM
2.8. REST Overexpression
Post-confluent 6 day serum-starved cells were trypsinized and placed in basic smooth muscle cell nucleofection solution (Amaxa) with Adenovirus containing REST (AdREST). Each sample contained 1 × 106 cells, 100 μL of nucleofection solution, and 600 pmol of siRNA. Samples were placed in electroporation cuvettes (Amaxa, Lonza, Basel, Switzerland) and nucleofected using program D-33 of the Amaxa nucleofector device. Following nucleofection, cells were immediately placed in RPMI media and incubated at 37°C and 5% CO2 for 15 min.
2.9. Chemotaxis
Smooth muscle migration was performed as previously [7]. Post-confluent day serum-restricted MASMCs were plated at 30,000–40,000 cells per well in the upper chamber of a 10 μm pore 96-well chemotaxis chamber (Millipore, MA, USA). The following solutions were placed in the lower chamber (diluted in serum-free media): vehicle, Platelet-derived Growth Factor (PDGF)-BB (30 ng/mL), and PDGF-BB + TRAM-34 (100 nM). The chamber was placed at 37°C overnight. Cells from the upper chamber were removed, and the filters were stained using the Diff-Quik Staining Kit (Fisher Scientific, Waltham, MA, USA). The migrated cells in a 40X field were manually counted.
2.10. Quantitative Reverse-Transcriptase PCR (qRT-PCR)
Quantitative reverse-transcriptase PCR (qRT-PCR) was performed as previously described [7,21,22]. Samples were quick frozen in liquid nitrogen and stored at −80°C until processed. Cultured cells were frozen in TRIzol solution. Total RNA was isolated according to the TRIzol published protocol. cDNA was transcribed from total RNA using High Capacity cDNA reverse transcriptase kit (Applied Biosystems 4368814, Foster City, CA, USA). A minus reverse transcriptase reaction was performed to ensure no genomic DNA contamination. Quantitative RT-PCR was performed on a MyiQ iCycler (Bio-Rad, model 170-9770). Each 20 μL reaction contained 1X Syber Green Master Mix (Bio-Rad, Hercules, CA, USA), 0.8 μM forward and reverse primers, and 1 μg of cDNA. Each reaction was initiated by a 95°C hold for 3 min in order to activate heat stable Taq polymerase, and reaction conditions were optimized for each set of primers. Target gene expression was normalized to 18S ribosomal RNA using the 2−ΔΔCT method [23].
2.11. Adenoviral Transfection and Migration Assay
Cells were serum-starved for 24 h, and then infected with adenovirus expressing the DNA-binding domain of REST (amino acid residues 234–437) inserted into pAdTrack-CMV [24] (AdREST, gift from Dr. Ian Wood) at an MOI of 100 plaque forming units for 24 h at 37°C. Cells were cultured in non-virus containing serum-free media for an additional 24 h and then plated at 30,000–40,000 cells per well in the upper chamber of a 10 μm pore 96-well chemotaxis chamber (Millipore). The following solutions were placed in the lower chamber (diluted in serum-free media): vehicle, PDGF-BB (30 ng/mL), and PDGF-BB + TRAM-34 (100 nM). The chamber was placed at 37°C for 4 h. Cells from the upper chamber were removed, and the filters were stained using the Diff-Quik Staining Kit (Fisher Scientific). The migrated cells in a 40X field were manually counted.
2.12. Statistical Analysis
All data are presented as mean ± SE. One-way ANOVA was used for all group comparisons, and significance was defined as p < 0.05. Data from the proximal, mid and distal segments were averaged for each animal unless a significant main or interaction effect of section was identified by initial two-way ANOVA.
3. RESULTS
3.1. Group Characteristics
Body weights for control and TRAM-34 treated mice were similar at the beginning (25.3 ± 0.6 g vs. 25.9 ± 0.4 g, resp.) and end (29.1 ± 1.0 vs. 28.8 ± 0.7 g, resp.) of the study (p > 0.05). Total cholesterol at the time of sacrifice was not affected by TRAM-34 (1455 ± 168 vs. 1334 ± 88 mg/dl, PO and TRAM, resp.).
3.2. Atherosclerotic Lesion and Carotid Artery Morphometry
We used the Partial Carotid Ligation (PCL) model in high-fat fed, Apoe−/− mice [12,13,16]. This model produces a low, oscillatory flow pattern (i.e. atheroprone with retrograde flow during diastole) in the left carotid artery (Figure 1b). In control animals, disturbed flow produced by PCL in the presence of a Western diet produced complex atherosclerotic lesions throughout the LCCA. Lesions consisted of fibrous, cellular matrix overlying lipid cores (Figure 2a). Conversely, TRAM-34 significantly reduced % stenosis (Figure 2b and 2c), medial area and Neointimal Size (NI; Figure 2d). Despite a greater plaque size in control, lumen area was comparable between groups due to a greater compensatory remodeling in control versus TRAM-34 as evident by a greater IEL and EEL area (Figure 2d). Compensatory (outward) remodeling is achieved by increasing vessel area (EEL area) in proportion to plaque size to maintaining luminal area. The slope of the relationship between total plaque burden (I + M area) and EEL area (i.e. remodeling index; Figure 2e) demonstrated a similar, linear response in both groups indicating that TRAM-34 reduces atherosclerotic plaque development without affecting normal compensatory remodeling.
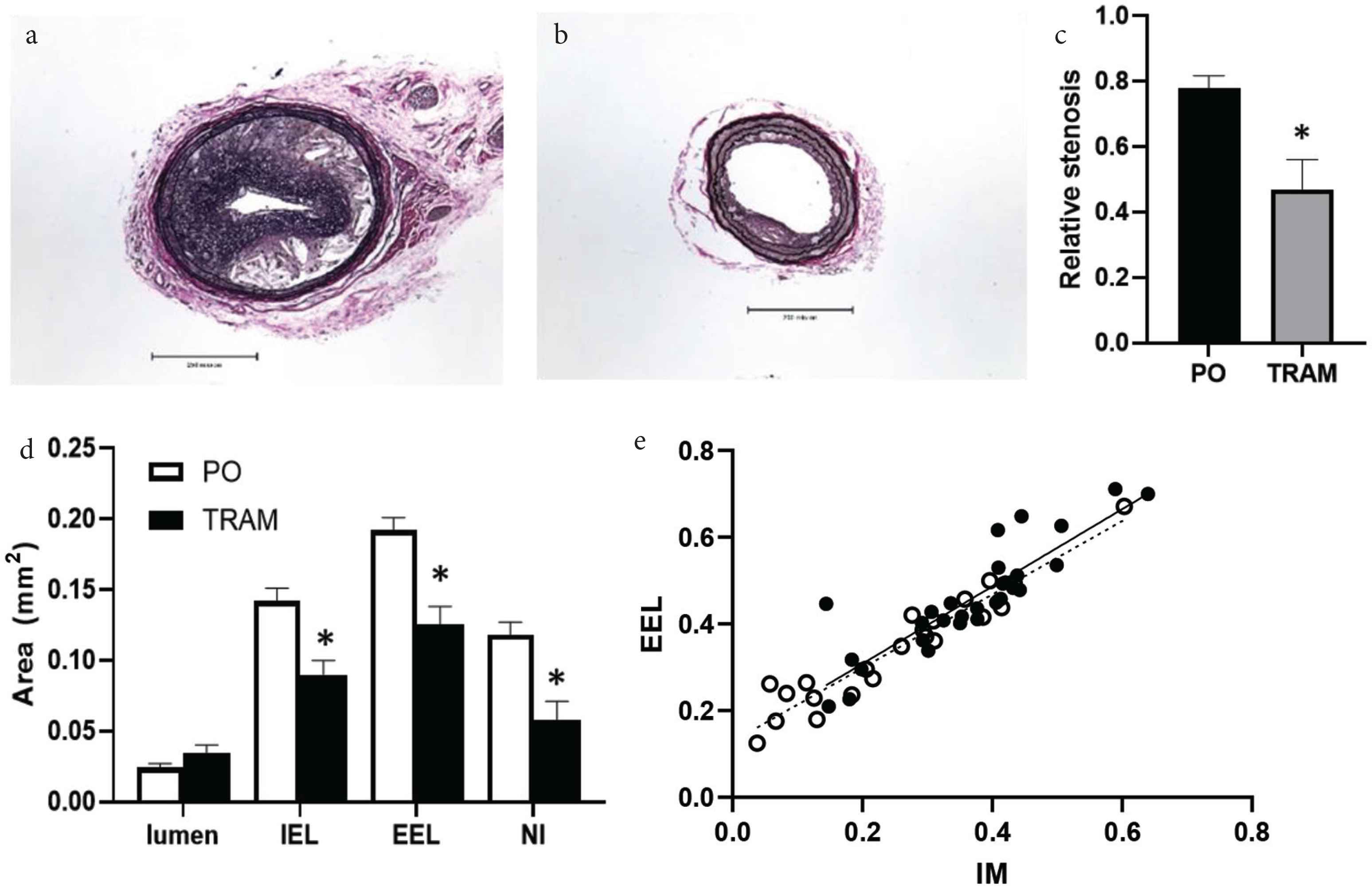
Representative Verhoeff-Van Gieson (VVG) stained LCCA sections from Peanut Oil (PO) vehicle (a) and TRAM-treated (b) mice. Scale bar = 500 μm. Morphometric data showing (c) reduced % stenosis and (d) internal and external elastic laminas (IEL and EEL), media and Neointimal (NI) areas in TRAM-34 treated mice (n = 11) vs. PO (n = 11), *p < 0.05. Relationship between EEL and lesion size (e) in proximal, mid and distal sections from PO (solid line) and TRAM-treated (dashed line) mice demonstrating compensatory remodeling was unaffected by TRAM-34.
3.3. TRAM-34 Alters Atherosclerotic Plaque Composition
Lesions from TRAM-34 treated mice versus controls contained significantly less collagen in the distal and proximal sections (Figure 3) and less fibronectin throughout the LCCA (Figure 4). The reason for the regional heterogeneity for collagen, i.e. lack of a reduction in the mid-section, is unknown. While there is some minor heterogeneity in lesion development along the ~700–1000 μm length of the LCCA, for all other endpoints measured there was no significant segmental difference. Thus, the overall major effect of KCa3.1 inhibition was a reduction in extracellular matrix content. Consistent with the role of intimal smooth muscle in matrix-production, total and relative smooth muscle content of the lesions was also significantly reduced by TRAM-34 (Figure 5). The majority of SMC within atherosclerotic lesions are of medial origin and migrate into the intima during lesion development [25–27]. The effect of KCa3.1 inhibition in reducing SMC content and associated matrix within the lesion suggests that KCa3.1 activation is involved in medial-to-intimal SMC migration. Previous studies by us and others [6,7,21] have shown TRAM-34 inhibits SMC migration in vitro. Consistent with this, we found that mouse aortic SMCs from Kcnn4−/− mice showed dramatically reduced chemotaxis to PDGF-BB (Figure 6). Thus overall, the lesions from TRAM-34 treated animals were smaller, yet with reduced relative extracellular matrix and smooth muscle content.
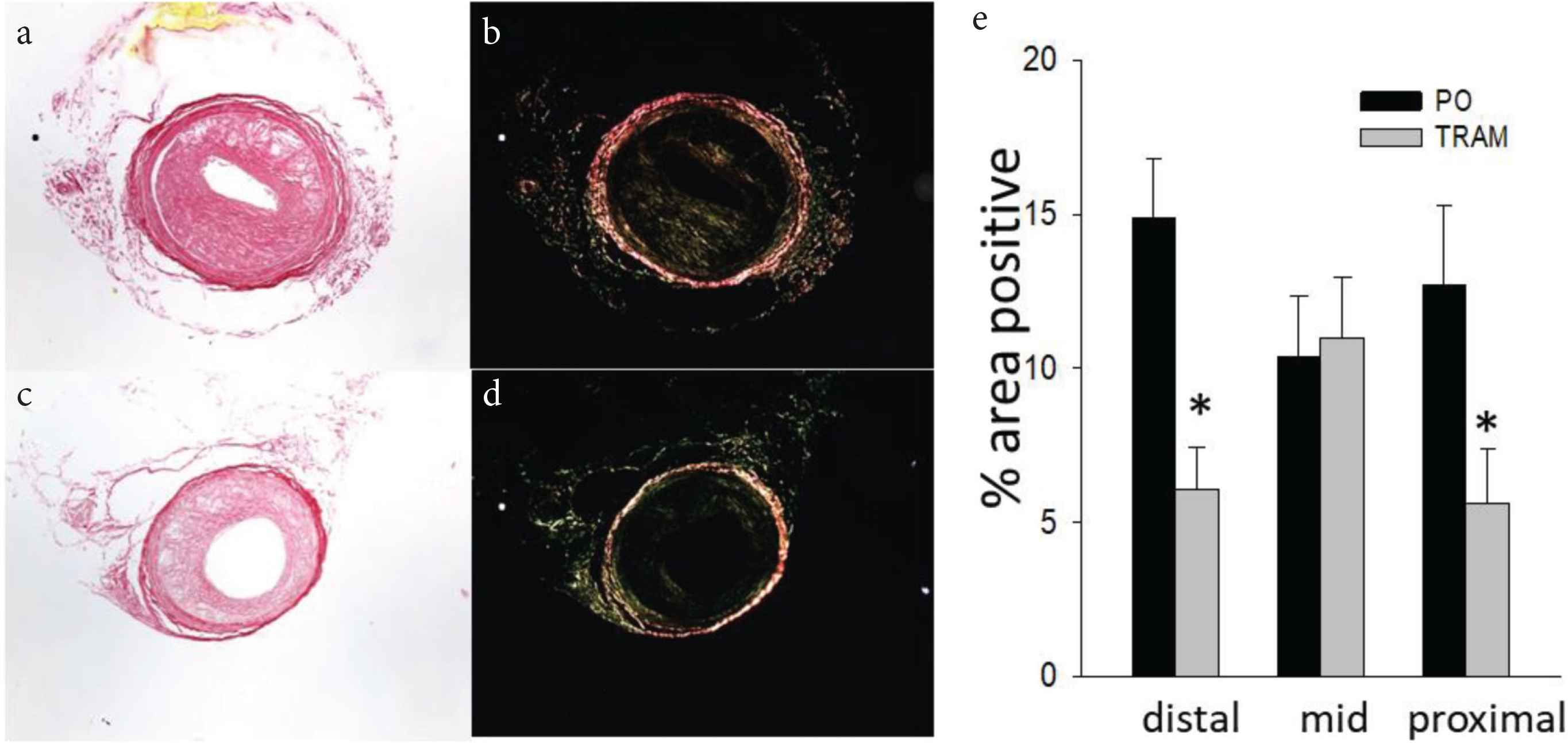
TRAM-34 reduces intimal collagen. LCCA sections from TRAM-34 (c and d) and Peanut Oil (PO; a and b) treated mice stained with picrosirius red under brightfield (a and c) and polarized (b and d) light. Mean ± S.E. for each carotid region (e); n = 10 and 11, for PO and TRAM, resp. *p < 0.05 vs. PO.
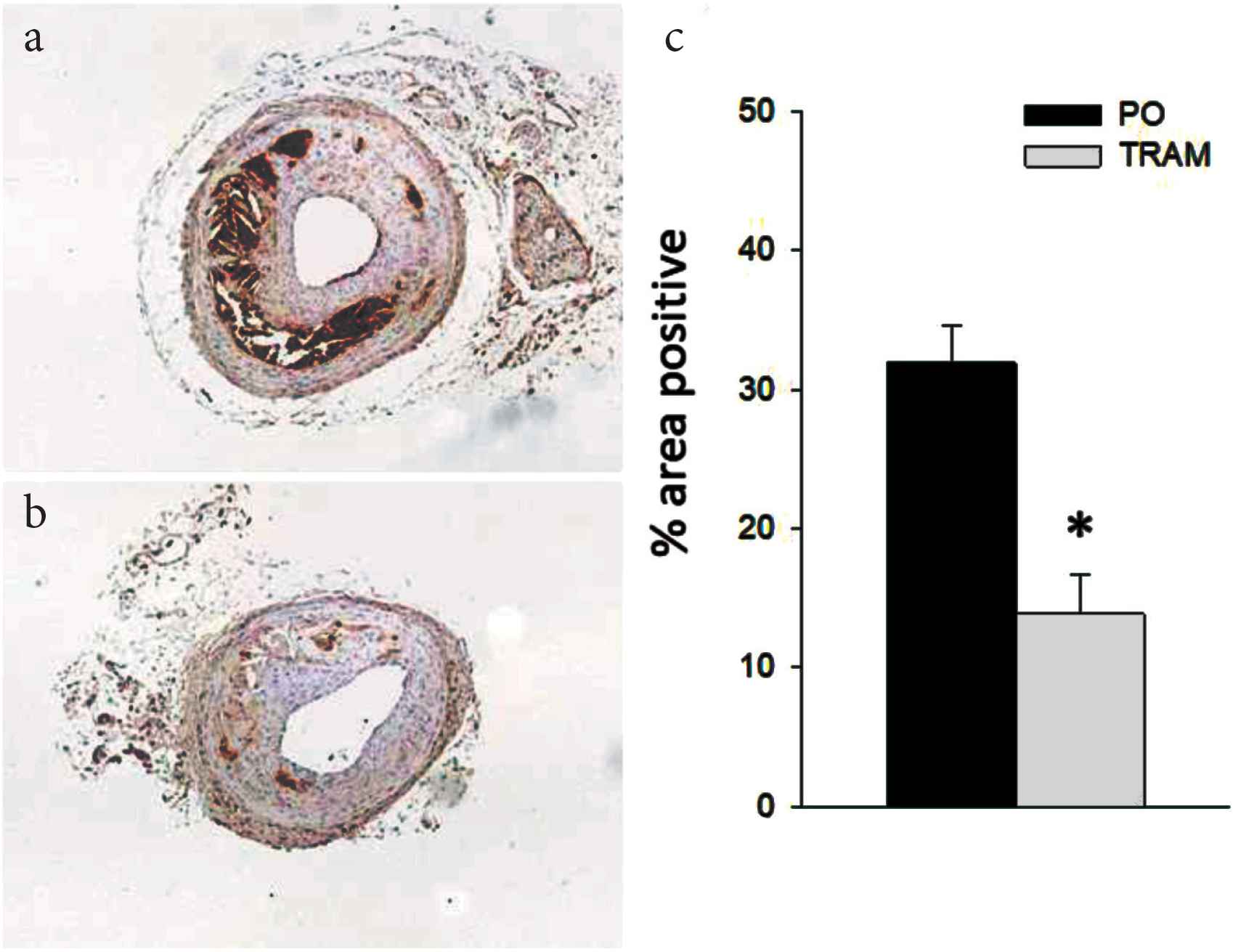
TRAM-34 reduces intimal fibronectin. Immunohistochemistry of LCCA sections from TRAM-34 (b) and Peanut Oil (PO; a) treated mice probed with anti-fibronectin. Mean ± S.E. for groups (c), n = 10 and 11, for PO and TRAM, resp. *p < 0.05 vs. PO.
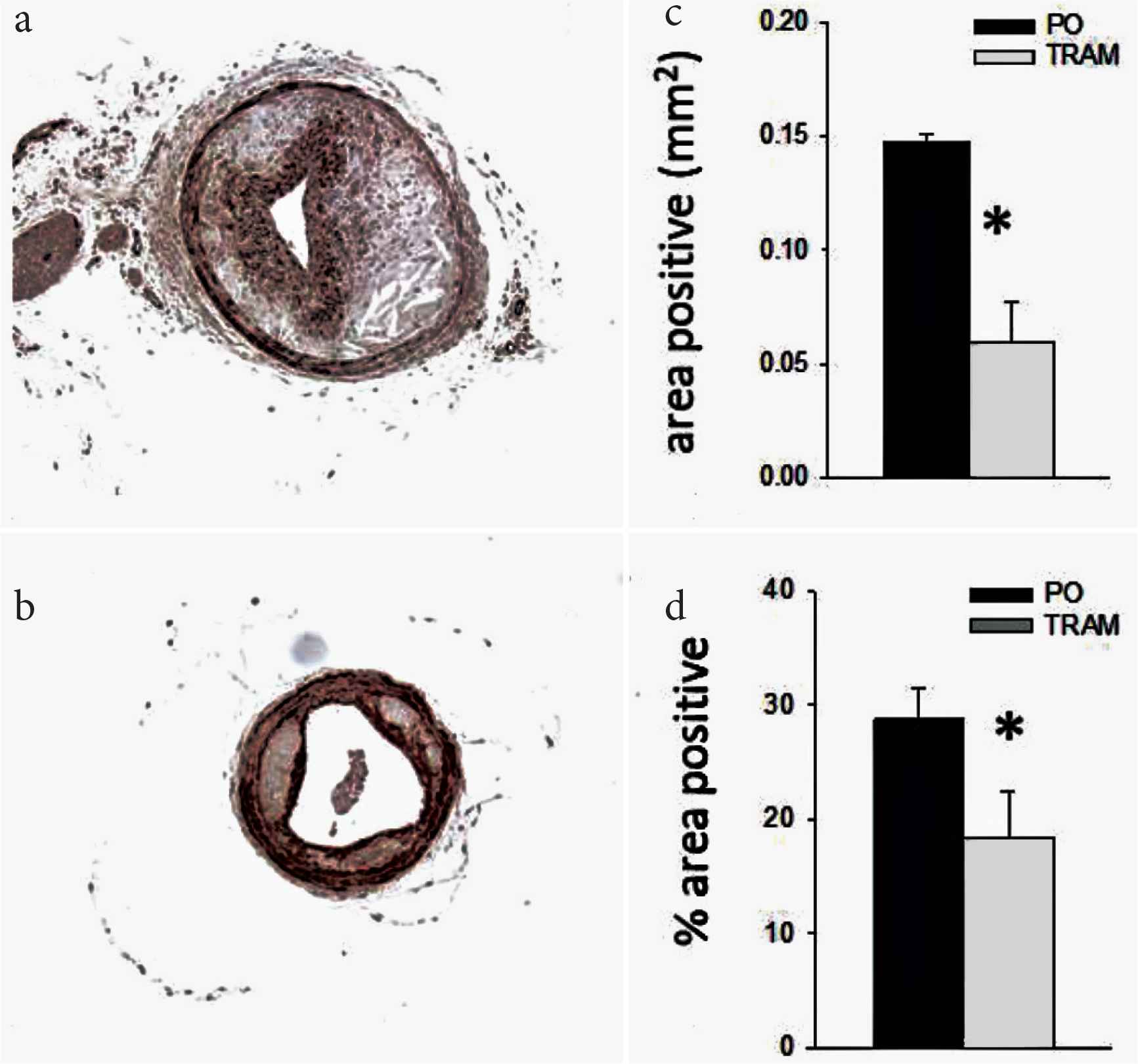
TRAM-34 reduces intimal smooth muscle content. Immunohistochemistry of LCCA sections from TRAM-34 (b) and Peanut Oil (PO; a) treated mice probed with anti-smooth muscle alpha actin (SMα A). Mean ± S.E. for groups for absolute (c) and relative (d) SMα A positive area, n = 10 and 11, for PO and TRAM, resp. *p < 0.05 vs. PO.
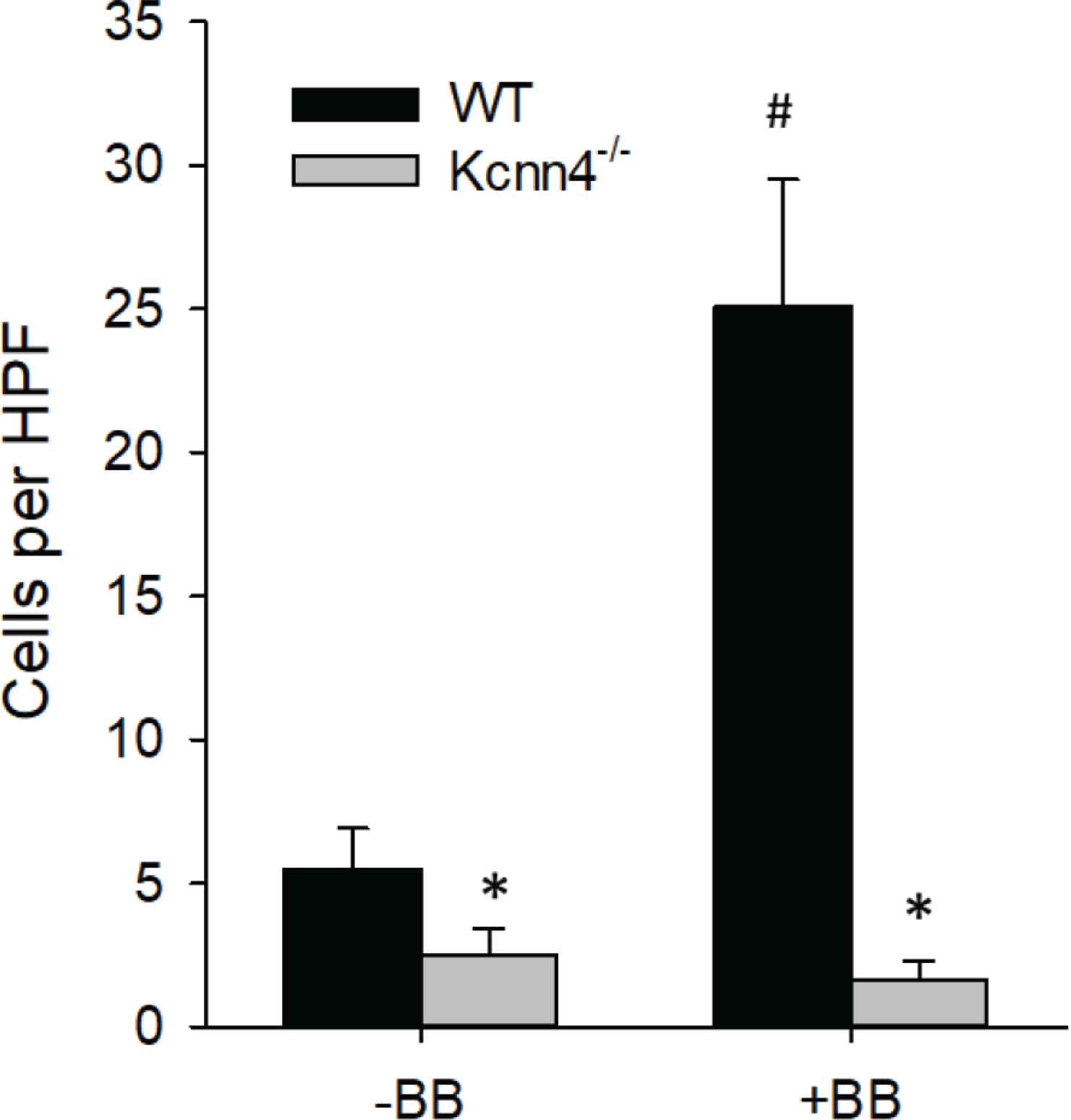
Genetic ablation of KCa3.1 inhibits migration in vitro. PDGF-BB induced migration in MASMC isolated from Kcnn4−/− mice is inhibited compared to Kcnn4+/+ (WT) cells. *p < 0.05 vs. WT; n = 8 per group.
Using a swine model of coronary restenosis, we have previously shown upregulation of KCa3.1 was associated with a decrease in the transcription factor repressor, REST in coronary arterial smooth muscle [4]. In the present study, loss of REST was also observed in medial SMC of mouse carotid arteries after partial carotid ligation (Figure 7). Downregulation of REST has been implicated as a mechanism for increased KCa3.1 expression and SMC migration. Consistent with this hypothesis, we found that transfection of MASMC with REST dramatically inhibits SMC migration (Figure 8). Furthermore, in cells overexpressing REST, inhibition of KCa3.1 with TRAM-34 had no further effect, consistent with REST suppression of KCa3.1-mediated migration.
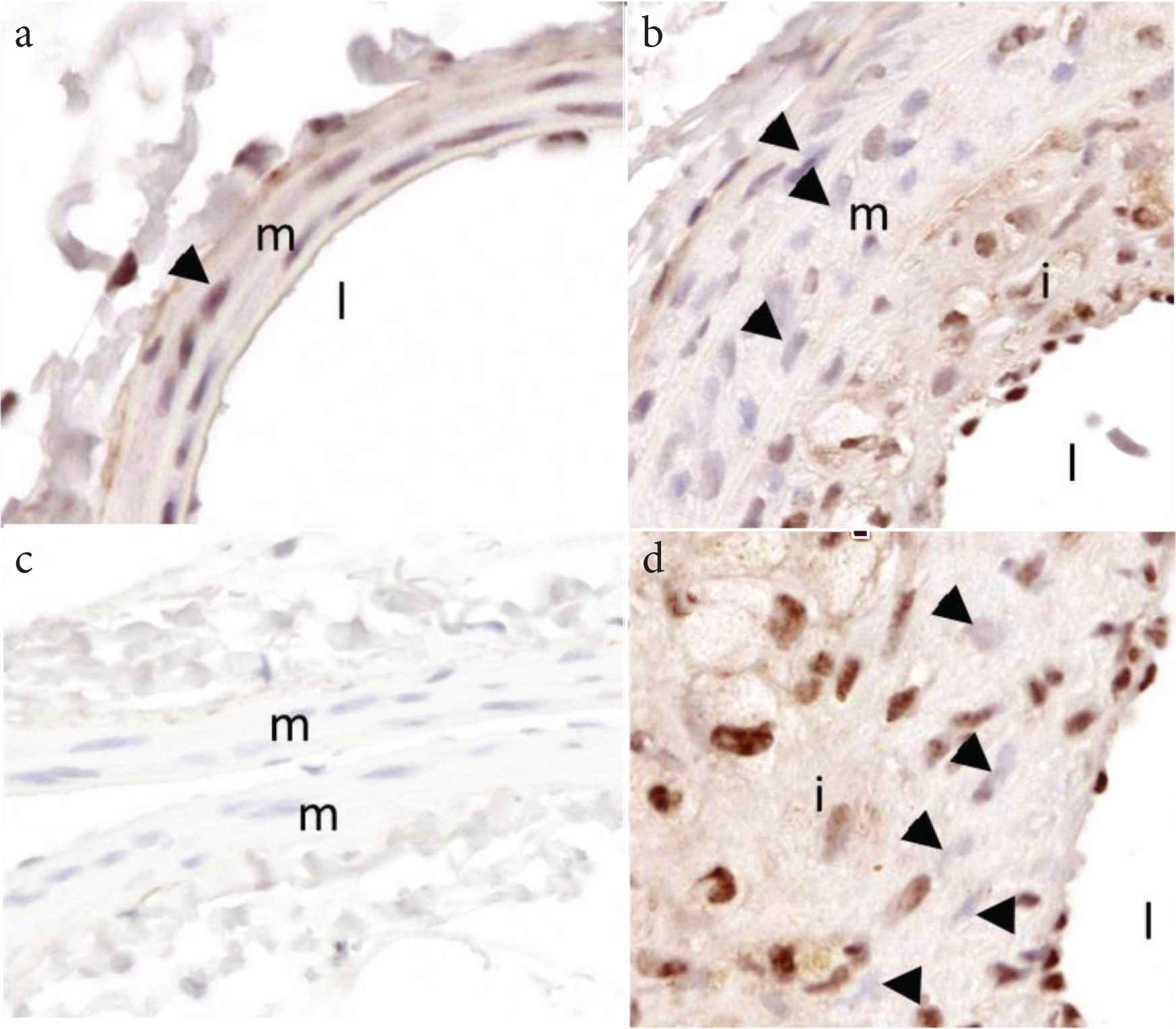
Representative images from an Apoe−/− mouse on Western diet demonstrating loss of medial SMC REST/NRSF in atherosclerosis. (a) Right common carotid artery (RCCA) showing nuclear staining for REST in medial SM (triangle). (b) Left common carotid artery (LCCA) after PCL showing loss of REST in medial SM (b) and intimal cells (d; triangles). (c) Non-immune control in RCCA. i, intima; m, media; l, lumen.
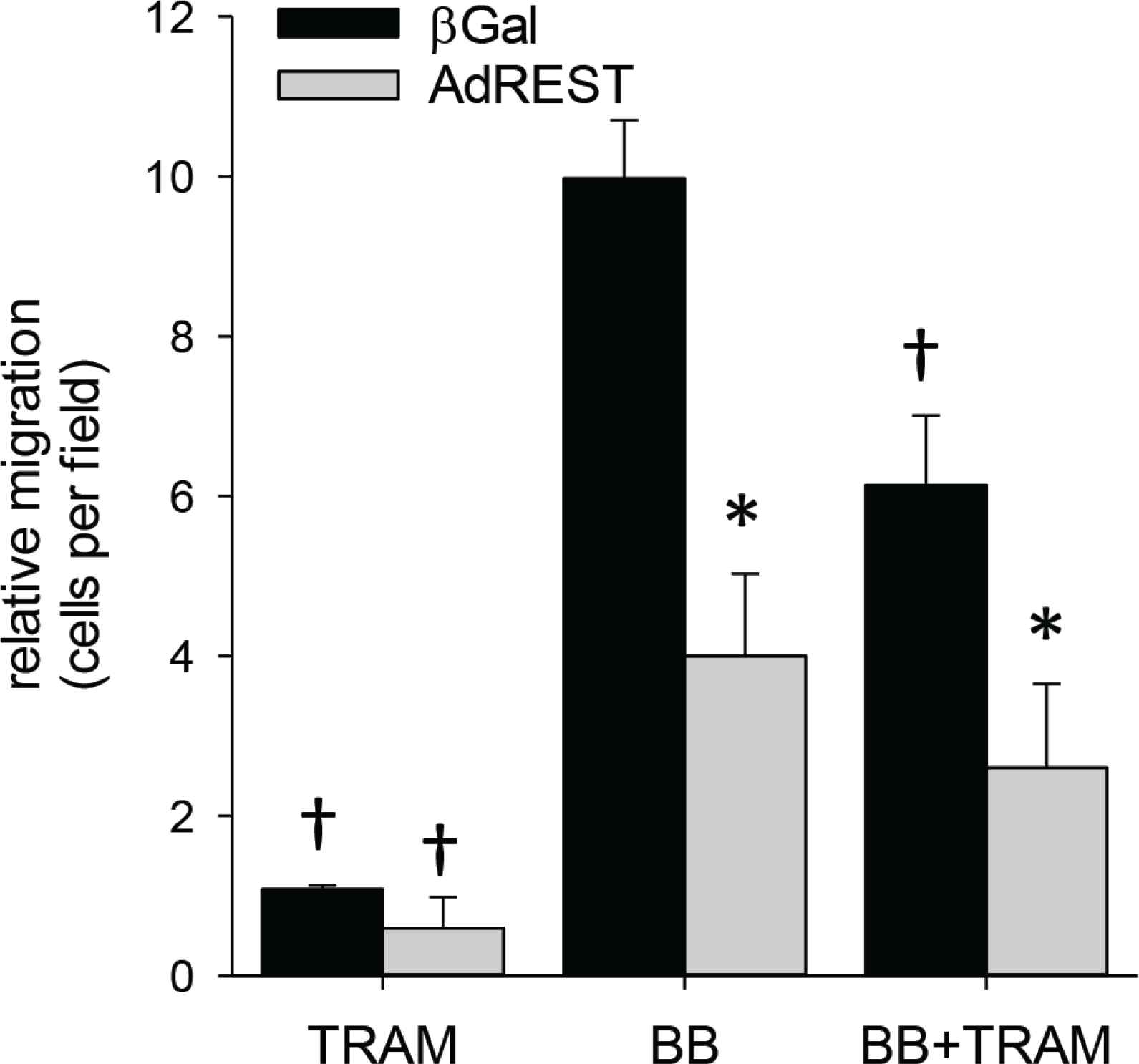
REST/NRSF inhibits migration. Ectopic overexpression of REST (AdREST) inhibits smooth muscle migration. Importantly, with REST overexpression, addition of TRAM produces no further inhibition, consistent with REST inhibition of KCa3.1. *p < 0.05 vs. β Gal, †p < 0.05 vs. BB; n = 7 per group.
4. DISCUSSION
The current study sought to determine if inhibition of KCa3.1 using the selective inhibitor TRAM-34 could reduce lesion development or alter composition in an in vivo model of atherosclerosis with compensatory remodeling. Atherosclerotic lesions are known to develop predominantly in regions of disturbed flow characterized by low, oscillatory shear in humans and animal models of atherosclerosis [10,11]. Toyama et al. [6] reported beneficial effects of TRAM-34 on lesion development in the aortic sinus. However, a limitation of this model is that it does not allow determination of potential effects on compensatory remodeling which can greatly influence plaque rupture probability [28,29]. Therefore, we used carotid partial ligation to produce low and oscillatory wall shear stress characteristic to induce complex lesion formation in an artery which exhibits compensatory remodeling [12–15]. Low and oscillatory wall shear stress promotes endothelial synthesis and release of potent mitogens, such as PDGF-BB, PDGF-AA [30] and endothelin (ET-1) [31] which stimulate SMCs to migrate from the medial into the intima through regionally disruptions in the IEL [32]. Importantly, we found that KCa3.1 inhibition with TRAM-34 reduced plaque volume, while simultaneously retaining appropriate compensatory remodeling. Maintaining compensatory remodeling is vital to potential therapeutic application of KCa3.1 inhibition as either failure to remodel or excessive expansive outward remodeling increases risk for plaque rupture [18].
Xu et al. [33] used combined partial ligation of the left renal and the LCCA and ECA using external collars in Apoe−/− mice. Similar to the current study, TRAM-34 reduced intimal area in the LCCA, however in contrast to our findings, they reported no change in intimal collagen content and an increased SMC content in carotid lesions. On the contrary, our finding of a reduction in SMC and collagen content in lesions is consistent with previous studies showing that TRAM-34 inhibition of KCa3.1 inhibits smooth muscle cell migration [6,7,21] and fibrosis [34–37]. Accordingly, one would expect a reduction in medial-to-intimal smooth muscle migration during lesion development with KCa3.1 inhibition and reduced intimal fibrosis. Partial renal artery ligation enhances vascular inflammation through elevations in circulating angiotensin II [38]. The enhanced macrophage infiltration, production of inflammatory cytokines and/or angiotensin II directly may account for the differing impact on lesion composition.
RE1-silencing transcription factor, also known as NRSF, is a transcriptional repressor of neuronal genes in non-neuronal cells that acts by binding to the consensus RE1/NRSE site [39]. Kcnn4, the gene encoding KCa3.1, contains a RE1/NRSE site and is inhibited by REST in VSM [4,40]. Accordingly, we and others have shown that REST expression is high in quiescent VSM and is decreased following injury during neointimal formation [4,40]. In the current study we similarly report reduced REST expression in SMC in the media underlying lesions and also demonstrated that overexpression of REST in smooth muscle inhibits migration in vitro in conjunction with KCa3.1 inhibition. These findings are consistent with a downregulation of REST leading to increased KCa3.1-induced SMC de-differentiation and medial-to-intimal migration.
In conclusion, our study provides further support for the important contribution of KCa3.1 activation in the progression of atherosclerotic lesion development and composition in response to low/oscillatory flow. Importantly, we found KCa3.1 inhibition had no adverse effect on compensatory remodeling, which alleviates a putative concern of therapeutic use of KCa3.1 inhibitors in atherosclerosis. Together with previous studies, these finding support the potential therapeutic application of pharmacological inhibition of KCa3.1 in limiting the progression of atherosclerosis. We also provide the first evidence supporting a role of REST in vascular smooth muscle in an in vivo model of atherosclerosis.
There are limitations to the current study. As noted, increased expression of the KCa3.1 has been observed in multiple cell types involved in atherosclerotic lesion progression including smooth muscle, macrophages and T-cells. Studies to date, including the current study, have relied on systemic pharmacological interventions in vivo. Genetic loss-of-function evidence of causality for KCa3.1 in plaque progression using in vivo models of atherosclerosis are lacking. Similarly, the relative contribution of KCa3.1 among cell types has not been determined and will require cell-specific, inducible silencing/overexpression to determine.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
DLT contributed to the conception and experimental design, collected and analyzed the data and wrote the first draft of the manuscript. DKB contributed to the conception and experimental design and final editing of the manuscript.
ABBREVIATIONS
- EEL,
external elastic lamina;
- IEL,
internal elastic lamina;
- KCa3.1,
intermediate conductance, Ca2+-activated potassium channel;
- Kcnn4,
gene encoding KCa3.1;
- LCCA,
left common carotid artery;
- MASMC,
mouse aortic smooth muscle cell;
- PCL,
partial carotid ligation;
- PDGF,
platelet-derived growth factor;
- RCCA-right
common carotid artery;
- REST/NRSF,
RE1-silencing transcription factor/neuron-restrictive silencer factor;
- SMC,
smooth muscle cell; VSM, vascular smooth muscle.
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - Darla L. Tharp AU - Douglas K. Bowles PY - 2021 DA - 2021/02/13 TI - KCa3.1 Inhibition Decreases Size and Alters Composition of Atherosclerotic Lesions Induced by Low, Oscillatory Flow JO - Artery Research SP - 93 EP - 100 VL - 27 IS - 2 SN - 1876-4401 UR - https://doi.org/10.2991/artres.k.210202.001 DO - 10.2991/artres.k.210202.001 ID - Tharp2021 ER -