
Interaction between the microcirculatory network and the systemic arterial pressure
- DOI
- 10.1016/j.artres.2010.11.002How to use a DOI?
- Abstract
The large arterial system and the microcirculatory network are generally studied as separated entities. We aimed to establish a link between the remodelling of microcirculatory and arterial blood pressure.
Experimental: Pre-hypertensive spontaneously hypertensive rats SHRs were maintained under hypoxic (12% O2) conditions for 8 weeks. The systolic blood pressure was lower by 26% in hypoxic vs. normoxic SHRs. Total peripheral vascular resistance was 30% lower in hypoxic than in normoxic SHRs. At the end of the hypoxic period, capillary density in skeletal muscle was 1.2-fold higher in hypoxic than in normoxic SHRs. Myocardial capillary density and VEGF-A protein contents were also 1.2- and 2.1-fold higher in hypoxic compared to normoxic SHRs. Thus, chronic normobaric hypoxia (1) activates VEGF-A-induced angiogenesis and thereafter (2) prevents or normalizes hypertension in the SHR.
Clinical: Increase in blood pressure (BP) is a hallmark of anti-angiogenic treatments. We used video microscopy to measure dermal capillary densities in the dorsum of the fingers. Measurements were carried out in 18 patients before and after 6-month treatment with bevacizumab. BP was increased compared with baseline from 129 ± 13/75 ± 7 mmHg to 145 ± 17/82 ± 7 mmHg. Compared with the baseline, mean dermal capillary density was significantly lower (75 ± 12 versus 83 ± 13/mm2). Thus, bevacizumab treatment resulted in capillary rarefaction, possibly participating to the rise in BP observed in most patients.
Thus, from experimental and clinical data, we might suggest that activation or blockade of angiogenesis may affect vascular resistance and, subsequently, blood pressure levels.
- Copyright
- © 2010 Association for Research into Arterial Structure and Physiology. Published by Elsevier B.V. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
The microcirculation is generally taken to include the smallest arteries, the arterioles, capillaries, and venules. Exchange of gases, nutrients, and metabolites between the blood and tissues occurs almost exclusively in the microcirculation, and adequate perfusion via the microcirculatory network is essential for the integrity of tissue and organ function. Therefore, functional and/or structural alterations in the microcirculation network could result in inadequate tissue perfusion and possible ischemic troubles. Actually, most of complications of cardiovascular diseases are related to insufficient tissue perfusion leading to ischemic troubles such as myocardial infarction, stroke, peripheral artery disease and ischemic nephropathy.
Both large arteries and microcirculation are extensively studied; however, there are generally considered as separated entities and believed to be poorly or not connected. In a series of animal experiments and clinical studies, we aimed to search for a link between the remodelling of microcirculatory and arterial blood pressure.
Microcirculation and arterial hypertension
In most forms of clinical and experimental hypertension, increased arterial blood pressure is associated with microvascular rarefaction and increased peripheral vascular resistances.1 Microvascular rarefaction, the reduced number or combined length of small vessels in a given volume of tissue, has been a consistent observation over many years in hypertensive patients and animal models.2 Two forms of rarefaction can be distinguished: the functional rarefaction, in which the number of perfused vessels is decreased without reduction of the number of vessels anatomically present, and the structural rarefaction, in which the number of vessels existing in the tissue is reduced. It has been suggested, initially on the basis of work in genetic spontaneously hypertensive rats (SHR), that functional rarefaction can progress to structural rarefaction: Prolonged vessel closure or non-perfusion can lead to its structural loss,3 analogous to the progression of active vasoconstriction to structural remodelling of resistance vessels. Both functional and structural rarefactions have been reported in skin tissue from hypertensive patients.4 Microvascular rarefaction has also been reported consistently in the myocardium, but here the situation is complicated by the fact that rarefaction may be due to actual loss of vessels or to failure of capillary growth to match the increase in myocardial tissue mass that usually accompanies hypertension.
Microvascular density might decrease because of either vessel destruction or insufficient angiogenesis, i.e., growth of new capillaries from pre-existing ones. This process proceeds during development and also in adults during physiological and pathological conditions. Oxidative stress and loss of NO-mediated vasodilatation may contribute to functional rarefaction. Oxidative stress and enhanced expression of both main enzymes that produce ROS in the microcirculation occur in SHR; furthermore, mice with genetically impaired NO signalling develop microvascular rarefaction. Apoptosis is also involved in microvascular rarefaction. Elevated levels of apoptosis have been observed in animal models of hypertension, and apoptotic endothelial cell death is involved in capillary structural rarefaction in glucocorticoid-induced hypertensive rats. Absence of flow induces apoptosis in endothelial cells adjacent to immobilized platelets and leukocytes, which may represent a mechanism linking functional and structural rarefaction. Apoptosis of a relatively small proportion of endothelial cells may be sufficient to mediate significant microvessel rarefaction.5 Apoptosis itself can be triggered by oxidative stress, and systemic application of antioxidants during growth reduces endothelial cell apoptosis and prevents development of microvascular rarefaction in SHR.
Cause and effect in hypertension
The cause-and-effect relationships of vascular rarefaction and hypertension are still debated. It is speculated that diffuse systemic rarefaction might be a primary defect in essential hypertension.6 There is evidence that experimental elevation of blood pressure causes an increase in generation of ROS in endothelial cells, which may trigger adverse functional and structural changes in microvessels.7,8 Such changes would be viewed as consequences of elevated pressure. However, there is considerable evidence that microvascular changes can also be a cause rather than a consequence of hypertension. In animal models of hypertension, increased ROS generation and arteriolar rarefaction occur even in parts of the vasculature not exposed to elevated blood pressure.9 Microvascular abnormalities occur early during development of hypertension in SHR, and prevention of oxidative stress by antioxidant treatment not only prevents rarefaction but also prevents the age-related development of hypertension. Furthermore, rarefaction occurs early in the development of human hypertension and has been detected in individuals with a familial predisposition to hypertension, even if they themselves are normotensive.10
Interestingly, epidemiological studies have shown that humans living at high altitude have lower systemic blood pressures than those living at the sea level.11–13 Although some metabolic or hormonal modifications have been described, the exact mechanism by which hypoxia prevents the development of hypertension remains largely undefined. A large prospective epidemiological study recently performed by the Swiss National Cohort Study Group reported results of mortality from a longitudinal, census-based record linkage study.14 Mortality data from 1990 to 2000, socio-demographic information, and places of birth and residence (men and women between 40 and 84 years of age living at altitudes of 259–1960 m) were collected. The 1.64 million German Swiss residents born in Switzerland provided 14.5 million person-years. Mortality from coronary heart disease and stroke significantly decreased with increasing altitude by 22% and 12% respectively per 1000 m. The authors suggested that protective effect of living at higher altitude on coronary heart disease and stroke mortality was consistent and became even stronger after adjustment for potential confounders. The effect was unlikely to be due to classic cardiovascular disease risk factors and rather could be explained by factors related to climate and/or altitude.
Experimental investigation
From this analyses of literature and using a physio-pathological point of view, we therefore hypothesized that hypoxia may trigger angiogenesis, resulting in reduction of blood pressure levels in spontaneously hypertensive rats.15 Indeed, hypoxia is known to trigger angiogenesis. The main mechanism of hypoxia-induced capillary growth involves the rise in hypoxia-inducible factor (HIF)-1 protein. HIF-1 binds to specific hypoxia-responsive element in the regulatory regions of several hypoxia-sensitive genes, such as vascular endothelial growth factor (VEGF)-A. VEGF-A is then secreted and binds to its cognate receptor tyrosine kinases, Flt-1 and Flk-1/KDR, located on the surface of vascular cells.
Five-week-old prehypertensive SHRs and age-matched normotensive Wistar–Kyoto (WKY) rats (n = 8 per group) were maintained under normobaric normoxic or hypoxic (12% O2) conditions for 8 weeks. Three weeks later, the systolic blood pressure was lower by 26% in hypoxic SHRs compared to normoxic SHRs (P < 0.05) and remained at the normoxic WKY level. Total peripheral vascular resistance, calculated as the mean arterial pressure/cardiac output (assessed by ultrasound imaging and Doppler), was 30% lower in hypoxic than in normoxic SHRs (P < 0.001) and returned to WKY levels. Interestingly, chronic hypoxia also significantly reduced systolic blood pressure in adult 12-week-old SHRs with established hypertension; blood pressure was normalized (versus normoxic WKY rats) after 4 weeks of hypoxia (Fig. 1). Changes in hemodynamic parameters were associated with activation of proangiogenic pathways. Protein levels of vascular endothelial growth factor (VEGF)-A in the skeletal muscles were increased by 2.2-fold in hypoxic compared to normoxic SHRs (P < 0.001, Fig. 2). At the end of the hypoxic period, capillary density in the quadriceps muscle was 1.2-fold higher in hypoxic than in normoxic SHRs (P < 0.001). Myocardial capillary density and VEGF-A protein contents (Fig. 3) were also 1.2- and 2.1-fold higher in hypoxic compared to normoxic SHRs (P < 0.001 and P < 0.05, respectively). Moreover, treatment with neutralizing VEGF-A antibody abrogated the hypoxia-induced angiogenesis and subsequently worsened arterial hypertension.
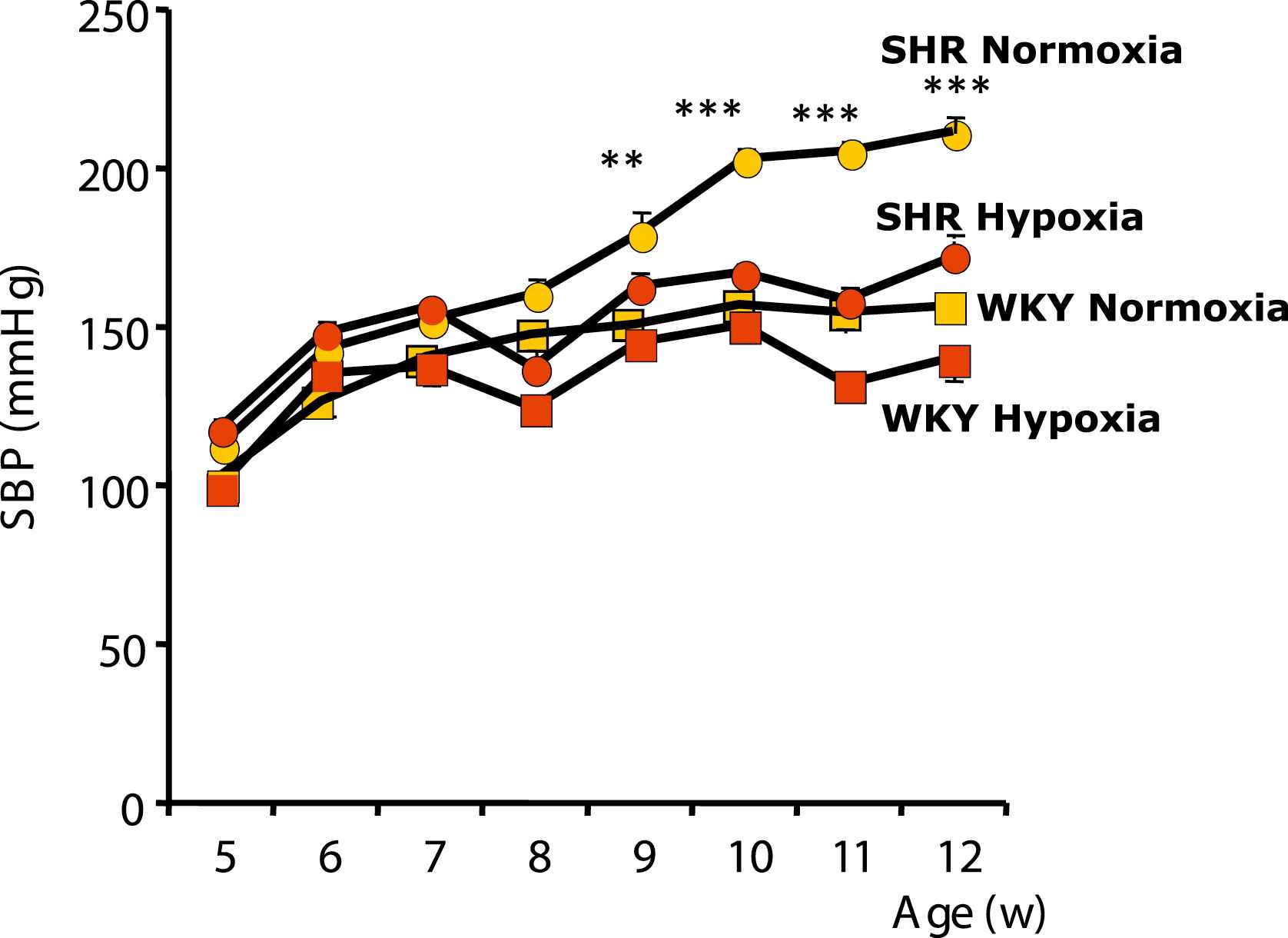
Systolic Blood Pressure (SBP, means ± SEM) in SHRs and WKY rats. A, Five-week-old rats were maintained for 8 weeks under normoxic or hypoxic conditions.*P < 0.05, ***P < 0.001.
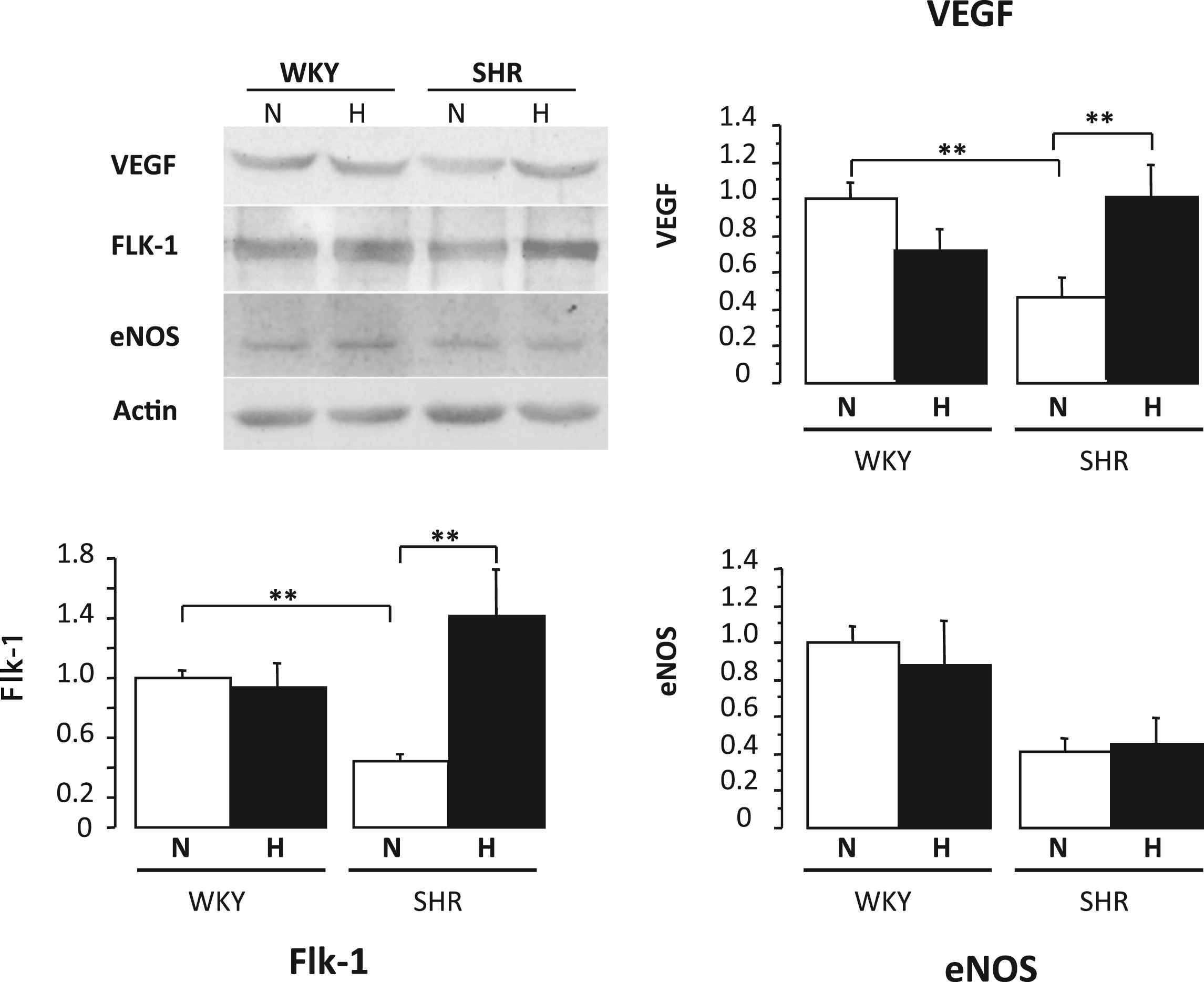
Quantitative evaluation of VEGF-A, Flk-1, and eNOS protein levels in the skeletal muscle (quadriceps) of WKY rats and SHRs under normoxic (N) or hypoxic (H) conditions. Values are means ± SEM. *P < 0.05, **P < 0.01.
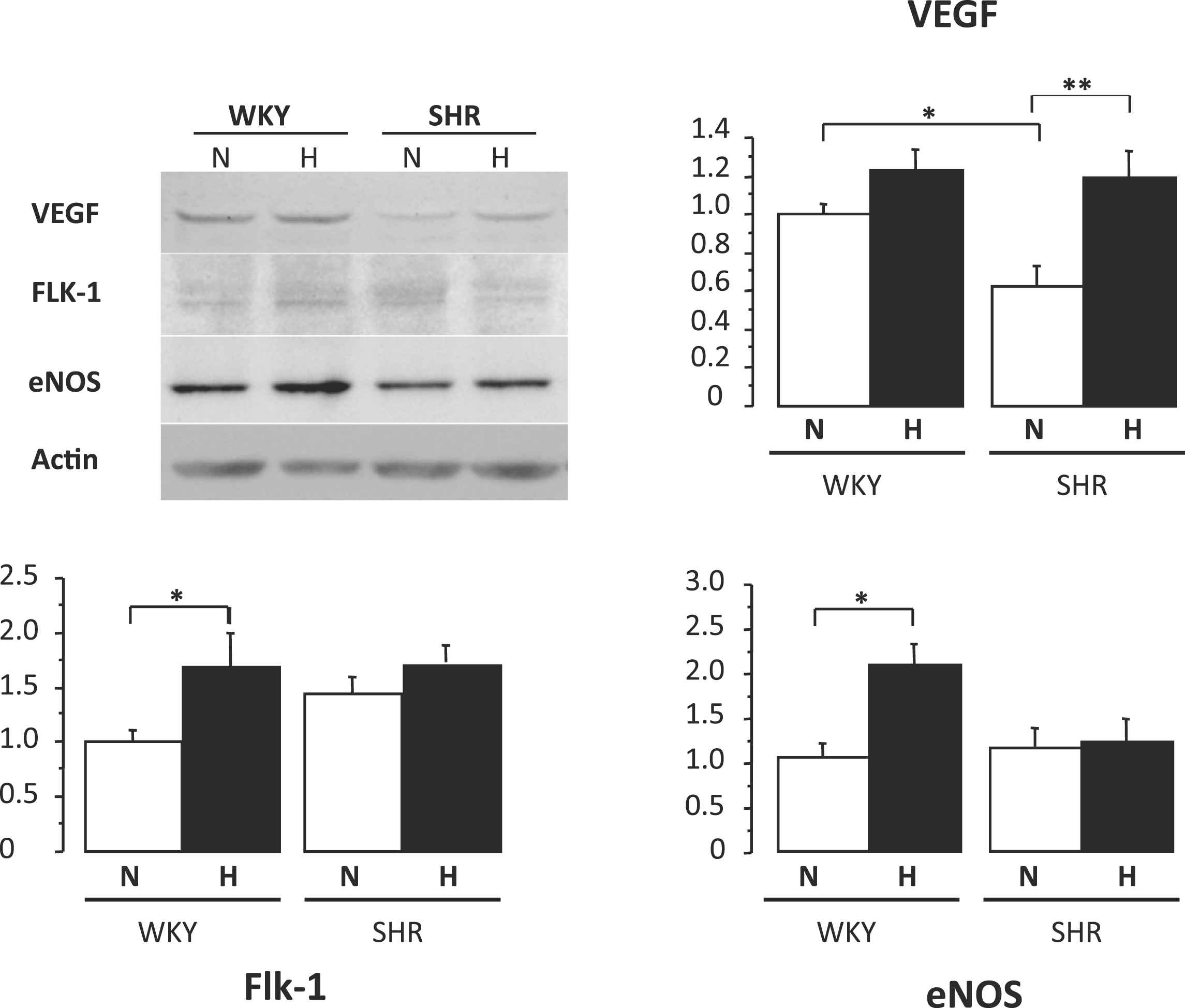
Quantitative evaluation of VEGF-A, Flk-1, and eNOS protein levels in the left ventricular myocardium heart of WKY rats and SHRs under normoxic (N) or hypoxic (H) conditions. Values are means ± SEM. *P < 0.05, **P < 0.01.
Therefore, our results suggest that chronic normobaric hypoxia
- -
Activates VEGF-A–induced angiogenesis and thereafter
- -
Prevents the occurrence of hypertension in young prehypertensive SHRs and,
- -
Normalizes blood pressure in adult SHRs with established hypertension.
The effect of hypoxia on blood pressure levels was likely related to activation of VEGFA–dependent angiogenesis. Hypoxia is a well-known stimulus for angiogenesis through activation of HIF-1 signalling. Although increased levels of HIF-1α mRNA have been reported, most of the studies suggest that HIF-1α is mainly regulated at the translational or posttranslational levels. However, HIF-1 α protein contents rapidly undergo proteosomal degradation. Accordingly, we did not find evidence of significant changes in HIF-1α mRNA levels. In contrast, we showed that hypoxia highly stimulates expression of HIF-1α related genes, VEGF-A, and its receptor, Flk-1. Activation of VEGF-A–related pathways has been shown to promote endothelial cells migration, proliferation, survival, and proteolytic activity and may thereby activate angiogenesis. Interestingly, in our experiments, co-treatment with VEGF-A–neutralizing antibody blocked hypoxia-related effects on angiogenesis and blood pressure levels. It is also noteworthy that VEGF-A protein contents were lower in normoxic SHRs than in normoxic WKY rats, suggesting that the decrease in VEGF-A levels could be involved in capillary rarefaction in this setting. In addition, others and we have previously shown that antihypertensive agents, such as angiotensin-converting enzyme inhibitor, raise VEGF-A levels and promote angiogenesis.16,17
Clinical study: blood pressure and pharmacological inhibition of angiogenesis
Arterial hypertension is a commonly reported side-effect in clinical trials testing all families of angiogenesis inhibitors, especially inhibitors of vascular endothelial growth factor (VEGF)/VEGFR-2 signalling.18 Whatever their initial level of blood pressure, every patient receiving anti-angiogenic treatment (bevacizumab, anti-VEGF antibody) evidence rapid and large increases in blood pressure.19 The same side effect was reported during treatment with anti-angiogenic drugs inhibiting the tyrosine kinase pathway.20 An incidence of 23% for all-grade hypertension and 6% for high-grade hypertension was noted with sorafenib (a small molecular inhibitor of several Tyrosine protein kinases (VEGF and PDGF) and Raf), with a six times greater relative risk for the development of all-grade hypertension compared with controls. Incidence and severity of hypertension as a toxic effect of sorafenib are very close to those reported with other anti-angiogenic molecules.
The mechanism underlying anti-angiogenic drugs-related hypertension is not yet clearly understood. As far as endothelial dysfunction and microvascular rarefaction are hallmarks in all forms of hypertension, we tested the hypothesis that anti-VEGF therapy could alter the microcirculation in nontumor tissues and, thus, result in an increase in blood pressure.
We used intravital video microscopy to measure dermal capillary densities in the dorsum of the fingers (Fig. 4). Capillary density was the mean of 4 fields measurements in a selected 3 × 3 mm area of the middle third of the phalanx. Measurements were carried out in patients before and after a 6-month treatment with bevacizumab.
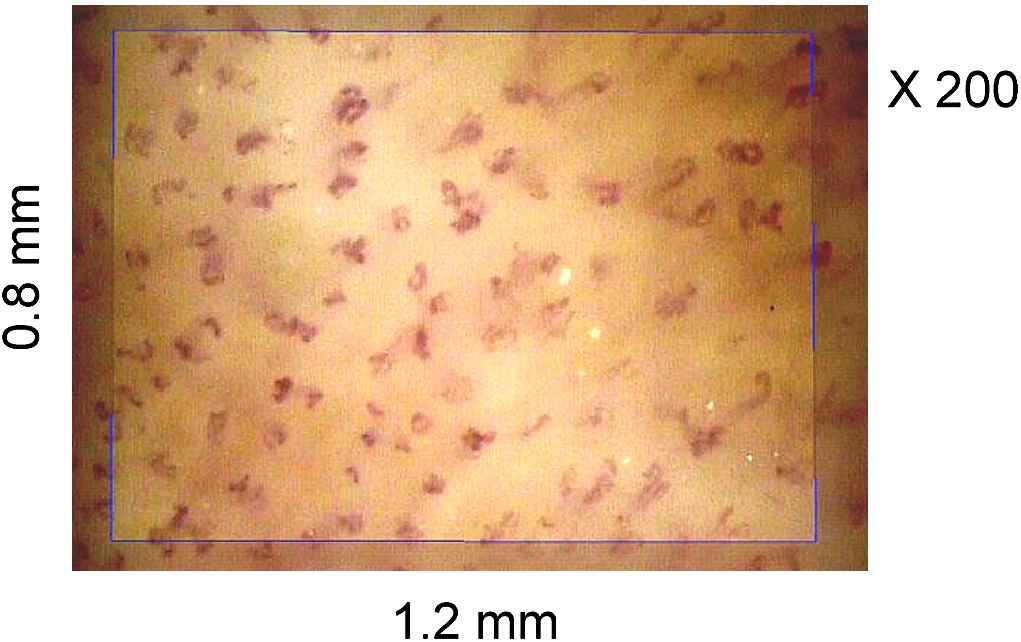
Typical video microscopic image of capillaries in the phalanx skin during venous occlusion. The rectangle represents a calibrated 1-mm2 surface. The structural capillary density is the number of capillary per surface unit.
Blood pressure was increased after 6 months of therapy compared with baseline, from 129 ± 13 mmHg (systolic pressure)/75 ± 7 mmHg (diastolic pressure) to 145 ± 17 mmHg/82 ± 7 mmHg for systolic and diastolic blood pressure, respectively (P < 0.0001). Compared with the baseline, mean dermal capillary density at 6 months was significantly lower (75 ± 12 versus 83 ± 13/mm2; P < 0.0001). Thus, we suggested that bevacizumab treatment resulted in arterial hypertension and capillary rarefaction; both changes closely associated.
This result has been recently confirmed21 in patients receiving telatinib, a small molecule angiogenesis inhibitor, after only a 5-week treatment period. Mean systolic and diastolic blood pressure values increased significantly at +6.6 mm Hg (P = 0.009) and +4.7 mm Hg (P = 0.016), respectively. Endothelial function was also altered by anti-angiogenic treatment: mean flow-mediated dilation value significantly decreased by −2.1% (P = 0.003). A statistically significant reduction of mean skin blood flux of 532% arbitrary units was seen (P = 0.015). Capillary density statistically significantly decreased from 20.8 to 16.7 capillary loops (P = 0.015). Proteinuria developed or increased in six patients during telatinib treatment. Thus, the increase in blood pressure observed in the treatment with telatinib may be caused by functional or structural microvascular rarefaction.
Finally, another group22 reported that sunitinib induced arterial hypertension and a significant decrease in capillary density; both parameters being negatively correlated.
All together, these results rise at least two points:
- 1.
Does angiogenesis occur in untreated healthy adults?
The answer is undoubtedly “Yes”; as shown in experimental in vivo models by Kamba et al.23 the maintenance of a normal capillary network is VEGF-dependent in several tissues. Capillaries of pancreatic islets, thyroid, adrenal cortex, pituitary, villi of small intestine, and epididymal adipose tissue are dependent on VEGF signalling for survival. The proportion of VEGF-dependent capillaries varies from organ to organ, with few being present in brain, retina, and heart. These findings indicate that capillaries in certain microvascular beds in the adult exhibit VEGF-dependent plasticity.
- 1.
Do changes in capillary density affect arterial pressure?
Our experimental data suggest that the hypoxic pathway, including HIF-1α, VEGF and VEGF receptors could modify, in parallel, microvascular density and blood pressure in hypertensives. Clinical data from patients receiving anti-angiogenic treatment seems to confirm the strong relationship between microcirculation, capillary density and blood pressure. This new concept needs further extensive investigation, especially in the clinical field.
References
Cite this article
TY - JOUR AU - Jose Vilar AU - Antony W. Kedra AU - Jean-Jacques Mourad AU - Jean-Sébastien Silvestre AU - Bernard I. Lévy PY - 2010 DA - 2010/12/02 TI - Interaction between the microcirculatory network and the systemic arterial pressure JO - Artery Research SP - 108 EP - 113 VL - 4 IS - 4 SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2010.11.002 DO - 10.1016/j.artres.2010.11.002 ID - Vilar2010 ER -